何首乌致肝损伤的信号通路及其作用机制
2024-03-28梁子晗李佳辉程爽袁卓雅荣文雅刘亚杰郝玉洁王睿林
梁子晗, 李佳辉, 程爽, 袁卓雅, 荣文雅, 刘亚杰, 郝玉洁, 王睿林
1 河南中医药大学第一临床医学院, 郑州 450000
2 南方医科大学中医药学院, 广州 510405
3 解放军总医院第五医学中心中医肝病科, 北京 100039
药物性肝损伤(DILI)是指由各类处方或非处方的化学药物、生物制剂、传统中药、天然药、保健品、膳食补充剂及其代谢产物乃至辅料等所诱发的肝损伤[1],较于其他肝损伤缺乏明显的特异性,具有潜在肝毒性,严重时可发展为肝衰竭甚至死亡。其中由中草药诱发的肝损伤具有临床使用广泛、潜伏期长、发病隐匿、诊断困难、机制复杂、难以预测等特点,占亚洲国家所有DILI发病率的25%[2],是医学界逐渐关注的重点问题。
中草药在我国已有上千年的应用史,自近代以来随着中草药致毒性事件频发,如何解决中草药毒性问题迫在眉睫。何首乌(polygonum multiflorum thunb,PM)作为传统常用补益类中草药,具有解毒、截疟、润肠通便的功效,经过炮制后可补肝肾、强筋骨、益精血、乌须发、化浊降脂,临床广泛用于治疗高脂血症、神经衰弱、老年痴呆、骨关节炎等疾病。研究[3]发现,PM主要含有二苯乙烯类、羟基蒽醌类衍生物、黄酮类、磷脂类等化合物,同时这些活性成分也具有潜在的肝毒性,如大黄素、二苯乙烯苷(TSG)、大黄酸等。继1988年PM引起肝毒性事件被首次报道后,美国、英国等国家也陆续出现服用PM导致肝损伤病例[4],此后多国发布PM及相关制剂警告及监管通告。因此,如何安全合理使用PM以减少和避免肝损伤发生是一项重要的临床问题。
1 PM致DILI机制简述
PM诱导的肝损伤(PM-DILI)以特异质型药物性肝损伤(IDILI)为主,但并不排除高剂量给药时诱发的直接肝毒性作用,其病理特征以肝细胞损伤型最为常见,其次是胆汁淤积型和混合型[5]。PM-DILI机制主要通过破坏线粒体功能,诱发免疫应激及氧化应激反应,改变胆汁酸稳态,干扰细胞色素P450酶(CYP450)、尿苷二磷酸葡萄糖醛酸转移酶(UGT)等相关代谢酶的表达等途径最终导致肝细胞死亡[6]。肝细胞死亡是PM-DILI的起始事件,主要包括凋亡、坏死、自噬等途径,其中坏死与凋亡是当前研究的主要热点。PM及其代谢产物作用于机体所激活的促死亡与抗死亡相关信号通路的平衡,决定着肝细胞死亡事件是否发生以及肝细胞损伤程度[7]。多种信号通路在PM-DILI发病过程中扮演着重要角色,并可能成为PM引起肝损伤的潜在治疗靶点。
2 PM通过线粒体相关信号通路致DILI机制
2.1 线粒体参与肝细胞死亡 线粒体作为细菌来源的细胞器,由围绕基质的两层膜组成,含有酶和线粒体DNA,是三磷酸腺苷(ATP)的主要部位,参与真核细胞的能量产生、代谢和凋亡,在调节各种药物诱导的肝细胞死亡中起核心作用[8]。线粒体毒性是导致DILI的主要机制之一,药物及其代谢产物经过多种化学反应,诱导细胞内应激,激活线粒体触发细胞死亡的外在与内在途径,通过相关信号影响Bcl2家族成员的平衡,如促凋亡(Bax、Bak)、抗凋亡(Bcl2、BclxL),进而影响线粒体的通透性[9](图1)。死亡受体激活或细胞应激使肝脏线粒体通透性转变(MPT),线粒体去极化,并导致ATP的严重消耗、细胞色素C释放、半胱天冬酶活化从而调节细胞程序性死亡[7]。由此可知,MPT在线粒体介导细胞死亡中发挥关键作用。药物及其代谢化合物积聚在线粒体,通过改变电子传递复合物的氧化还原状态,或与这些复合物中的蛋白质共价结合,共同导致电子流中断促进活性氧(ROS)的产生,进一步损害呼吸链,导致更多的ROS产生,当ROS达到一定阈值时,激活关键的c-Jun N端激酶(JNK)信号通路,同时导致MPT[7]。由此可见,药物及其代谢产物作用于线粒体通过抑制其呼吸链、增加ROS以及MPT最终导致DILI的发生(图2)。

图1 线粒体介导的细胞死亡机制[9]Figure 1 Mitochondria-mediated cellular death mechanism[9]
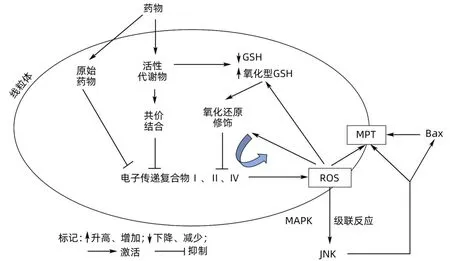
图2 线粒体介导的DILI机制[7]Figure 2 Mechanism of mitochondria-mediated DILI[7]
2.2 JNK信号通路 JNK信号通路是丝裂原活化蛋白激酶蛋白家族成员,主要由细胞因子和环境应激激活,参与细胞增殖、分化、发育、炎症反应与凋亡[10]。线粒体是JNK促凋亡信号传导的主要靶点,Han等[7]研究发现,JNK信号通路介导线粒体在对乙酰氨基酚诱导的DILI中起重要作用。在肝毒性剂量下,对乙酰氨基酚及代谢产物与线粒体中的谷胱甘肽(GSH)和蛋白质共价结合,导致肝细胞发生剧烈的氧化还原变化,GSH过氧化物酶消耗GSH解毒导致线粒体中H2O2释放增加,H2O2是ROS最主要成分,其含量的增加是细胞质中JNK活化的关键因素。JNK激活后可易位到线粒体,诱导MPT的发生及细胞色素C、凋亡诱导因子等凋亡因子的释放,抑制线粒体生物能量产生,从而促进肝细胞坏死与凋亡[7]。JNK激活后,本身也可通过线粒体ROS生成,增加导致细胞凋亡信号调节激酶1的激活,从而实现自我放大、自身激活。与此同时,JNK还可以直接参与Bcl-2家族成员的调控,如激活Bax、使Bcl-xl失活[11]。研究[12]发现,PM可降低肝线粒体琥珀酸脱氢酶活力和呼吸链复合物Ⅰ活性,影响还原型辅酶Ⅰ呼吸链功能,导致细胞代谢障碍,从而增加ROS生成,激活JNK;此外,PM可调控Fas/FasL死亡受体通路,上调促凋亡因子Bax表达,下调抗凋亡因子Bcl-2表达,影响肝脏线粒体的通透性。Lin等[13]研究发现,大黄素可通过抑制线粒体呼吸链复合物功能,影响氧化磷酸化途径,导致ROS生产增加,随后激活JNK,诱导肝细胞死亡。研究[14-15]发现,PM可降低肝细胞中GSH水平,提高ROS含量,增强JNK表达水平。以上研究提示,PM可能通过影响线粒体呼吸链功能,改变线粒体通透性,消耗GSH,增加ROS含量,激活JNK信号通路,从而诱导DILI的发生。
2.3 ROS/ATM/P53通路 毛细血管扩张性共济失调突变基因(ATM)编码的蛋白是一种丝氨酸/苏氨酸激酶,在DNA双链断裂时被激活,通过磷酸化参与DNA修复和细胞周期控制的关键底物来响应DNA损伤[16]。P53作为肿瘤抑制基因,参与调节细胞周期检查点、细胞凋亡、衰老、DNA修复,维持基因组完整性和控制血管生成[17]。Lai等[18]研究发现,大黄素可触发ROS产生并破坏线粒体膜电位,使ATM在Ser1981位点磷酸化激活,伴随着P53在Ser15位点磷酸化激活,Bax表达增强,通过线粒体介导细胞色素C释放诱导细胞凋亡。Bounda等[19]研究发现,大黄酸通过线粒体途径诱导细胞凋亡的机制与上述过程相似。以上研究提示,PM活性成分可能通过激活ATM、P53,增强Bax的表达,通过线粒体介导与其他危险因素共同参与DILI的发生。
3 PM通过法尼醇X受体(FXR)相关信号通路致DILI机制
3.1 FXR维护胆汁稳态 FXR作为一种胆汁酸激活的转录因子,可感知胆汁酸水平,对维护胆汁酸稳态至关重要。当胆汁酸水平升高时,FXR将被激活,诱导抑制肝细胞胆汁酸生物合成的程序[20]。胆汁酸是FXR的生理配体,FXR与胆汁酸结合后,可抑制胆汁酸合成中的限速酶细胞色素P450家族成员7A1(CYP7A1),同时激活回肠胆汁酸转运蛋白,胆汁酸通过与FXR结合的方式调节自身的合成和运输。与此同时,FXR还可以调节胆盐输出泵(BSEP)、肝脏多药耐药相关蛋白2(MRP2)、钠牛磺胆酸共转运多肽(NTCP)等相关基因,调控胆汁酸的摄取、转运与排泄[21](图3)。Sinal等[22]研究发现,在饮食中添加1%的胆酸会导致FXR基因敲除小鼠血清胆汁酸水平升高约8倍。由此可见,FXR信号在胆汁淤积性肝损伤中发挥重要的保护作用。
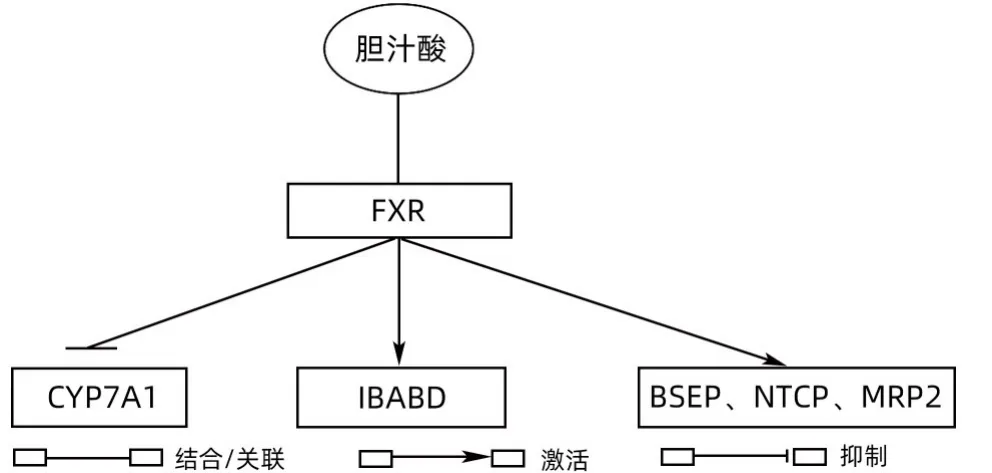
图3 FXR维护胆汁稳态机制Figure 3 Mechanism of FXR-maintained bile homeostasis
3.2 腺苷酸活化蛋白激酶(AMPK)/FXR通路 AMPK是一种能量传感器蛋白激酶,在调节细胞能量代谢中起关键作用,是FXR转录活性的负调节因子,BSEP作为FXR的直接靶基因,由FXR激活,是人类主要的管状胆盐出口泵,通过以胆盐依赖性方式调节胆汁脂质分泌[23]。Li等[24]研究发现,二甲双胍可通过激活AMPK来加重α-萘基异硫氰酸盐(ANIT)诱导的胆汁淤积,同时抑制FXR表达。Lien等[25]发现熊去氧胆酸通过抑制AMPK减轻雌激素诱导的胆汁淤积。大黄素是已知的AMPK激活剂,PM大黄素可通过AMPK抑制FXR的激活,下调BSEP等相关基因表达,加重胆汁淤积[26]。孙萌等[27]发现PM TSG呈剂量依赖性下调FXR表达,并抑制下游靶点BSEP、CYP7B1的表达。以上研究提示,PM活性成分可能通过激活AMPK,抑制FXR,减少BSEP,下调CYP7B1表达,从而具有诱导肝内胆汁淤积加重肝细胞损伤的潜在风险。
3.3 FXR/小异二聚体伴侣(SHP)通路 孤儿核受体SHP是胆汁酸的关键调节因子,其被FXR诱导激活后与孤儿核受体肝受体同系物-1结合,可抑制CYP7A1的表达,从而减少胆汁酸的生成,维护胆汁稳态[28]。此外,人成纤维细胞生长因子19(FGF19)的磷酸化可显著增强SHP的基因功能,FGF19是FGFR4的高亲和力肝素依赖性配体,被FXR激活后,通过肝肠循环从回肠分泌到肝脏,结合并激活FGFR4受体,与β-Klotho组成复合物,进一步激活SHP等信号通路,共同抑制CYP7A1的表达[29]。由此可见,FXR/SHP信号通路在调控肝肠循环胆汁酸水平中发挥着重要作用。Zhang等[30]研究发现,PM可通过FXR/SHP通路诱导IDILI的发生。Dai等[31]、韩宗萍等[32]研究发现PM可抑制FXR的表达,下调SHP、CYP7A1的表达。Sun等[33]研究发现PM TSG可抑制FXR激活,下调SHP表达,干扰胆汁酸合成的替代途径,加重胆汁淤积。以上研究提示,PM可能通过抑制FXR/SHP信号通路导致胆汁淤积造成肝细胞损伤。
4 PM通过Toll样受体4(TLR4)相关信号通路致DILI机制
4.1 TLR4参与免疫应激调控 TLR4是一种Ⅰ型跨膜蛋白,在识别病原体和激活先天免疫中起关键作用,主要在抗原呈递细胞(如巨噬细胞等)上表达,细菌脂多糖(LPS)是其主要配体[34]。LPS是免疫系统中强有效的诱导剂,肝脏受损后其水平明显增加,与TLR4结合后可通过髓样细胞分化因子88(MyD88)依赖途径和MyD88非依赖途径进行信号传导,增加炎性介质的释放,并激活机体免疫炎症反应[35]。研究表明,LPS介导的TLR4通路在肝损伤过程中发挥重要作用。
4.2 TLR4/MyD88/核因子κB(NF-κB)通路 MyD88作为一种胞质适配器蛋白,在先天性和适应性免疫反应中起核心作用,是TLR4和IL-1受体(IL-1R)的关键下游靶点蛋白[36]。TLR4与LPS结合后,发生二聚化,并传导至胞内,与MyD88相互作用,结合并磷酸化IL-1R相关激酶,随后激活肿瘤坏死因子α受体相关因子6(TRAF6),进而激活NF-κB诱导激酶,使IκB激酶磷酸化,IκB泛素化降解后释放NF-κB进入细胞核,引起TNF-α、IL-1ß、IL-6等相关炎症介质的释放,最终导致组织、器官的炎症与损伤[37]。谢丽华等[38]研究表明经LPS诱导后,PM醇提液可上调mTLR4表达,影响MyD88及下游通路的表达,且不呈剂量依赖性。潘韵铮等[39]发现LPS与顺式TSG联合应用可显著增强肝脏TLR4、MyD88、NF-κB的表达,提高血液中IL-1β、TNF-α等炎性因子的水平。Long等[40]研究发现,PM可激活TLR4,选择性上调MyD88表达,诱导下游NF-κB信号通路。另有研究[41]发现,TLR4/NF-κB通路是顺式TSG导致IDILI的重要基因通路。以上研究提示,PM可能经LPS诱导激活TLR4/MyD88/NF-κB信号通路,参与机体免疫应激,导致IDILI的发生。
4.3 TLR4/β干扰素TIR结构域衔接蛋白(TRIF)/干扰素调节因子-3(IRF-3)通路 TRIF作为Toll/IL-1受体同源结构域的衔接蛋白,过表达时可激活NF-κB,是比MyD88更有效的IFN-β诱导剂,在LPS激活IRF3和诱导IFN-β中起关键作用。TRIF与TLR4被相关桥接分子(如易位关联膜蛋白62、TLR衔接分子-2等)连接,共同导致IRF3易位到细胞核,通过TRAF6相关途径激活NF-κB[42]。毛宏梅等[43]、谢丽华[38]研究发现PM醇提液经LPS诱导可显著提高肝细胞TLR4、TRIF和IRF-3 mRNA和蛋白表达水平。由此可见TLR4/TRIF/IRF-3信号通路可能是LPS介导PM致IDILI作用机制之一。
5 PM通过PRKR样内质网激酶(PERK)/转录激活因子4(ATF4)/增强子结合蛋白同源蛋白(CHOP)通路致DILI机制
肝脏暴露于生物和化学刺激时需要通过内质网应激、自噬与氧化还原应激来维持自身细胞与组织稳态。内质网主要调节细胞内Ca2+储存及稳态、蛋白质折叠加工。当机体受到外界因素刺激发生氧化应激、炎症、钙代谢紊乱等事件后,导致错误折叠与未折叠蛋白质蓄积于内质网,细胞内平衡紊乱引发内质网应激,通过转录因子如CHOP等途径诱导细胞自噬与凋亡[44]。研究[45-46]发现,由内质网应激介导的细胞凋亡与肝脏疾病的发生密切相关。PRKR是Ⅰ型内质网膜蛋白,参与综合应激反应和未折叠蛋白反应。当发生内质网应激后,激活PERK,磷酸化真核翻译起始因子2α(eIF2α),随之激活ATF4,导致CHOP过度表达,通过增加p-eIF2α去磷酸化、提高Bax水平、加强内质网氧化酶1α表达、抑制抗凋亡蛋白激酶B活化等途径促进细胞凋亡[47]。研究[48]发现,PM大黄素经代谢酶活化后可产生肝毒性更强的5-羟基大黄素,其可通过破坏L02细胞线粒体结构和功能,增加ROS产生,致使胞内Ca2+超载从而激活内质网PERK/ATF4/CHOP信号通路,从而增加DILI发生风险。
6 PM通过过氧化物酶体增殖激活受体γ(PPARγ)信号通路致DILI机制
PPARγ是脂肪细胞分化的调节剂,主要分布在肝脏与脂肪细胞中,参与脂质和葡萄糖代谢、细胞增殖和凋亡以及免疫调节[49]。Li等[50]研究发现,PPARγ激活后可通过增强IκB-α表达,减少NF-κB核易位,降低NF-κB的DNA结合活性,从而减少细胞因子的产生和组织损伤。研究[51]表明,经LPS/顺式TSG处理的大鼠,p65水平升高,IκB-α和PPARγ表达降低,且PPARγ通路与LPS/顺式-SG诱导的肝损伤呈负相关,加用吡格列酮(PPARγ受体激动剂)处理后可增加PPARγ和IκB-α的蛋白质水平。Meng等[52]研究发现,PM顺式TSG可下调PPARγ表达,激活NF-κB信号通路,增加TNF-α、IL-6等促炎细胞因子诱导IDILI的发生。以上研究提示,PM可能通过抑制PPARγ相关信号通诱导IDILI的发生发展。
7 PM通过其他相关通路致DILI机制
磷脂酰肌醇3激酶(PI3K)是由调节亚基p85和催化亚基p110构成二聚体,当其与生长因子受体结合后,可活化丝氨酸/苏氨酸激酶(Akt),随后激活或抑制相关下游蛋白(如Caspase-9),从而发挥促细胞生存、增殖并抑制细胞凋亡及调控葡萄糖代谢等作用[53]。研究[54]发现,PM可通过降低转运蛋白溶质载体家族16成员2表达致使PI3K/Akt信号通路无法顺利激活,进而导致促凋亡蛋白及因子(如P53蛋白、Caspase-9)无法被抑制而导致肝细胞凋亡。核因子红细胞2相关因子2(Nrf2)/Kelch样ECH关联蛋白1(Keap1)信号通路是一种细胞内防御机制,参与细胞抵消氧化应激,而PM在致死毒性剂量下可抑制肝细胞Nrf2信号通路[55];Gonzalez等[56]研究发现CYP2E1可激活CHCl3、CCl4等具有肝毒性的化学物质,产生ROS导致肝细胞毒性加重。汪美汐等[57]研究发现PM大黄素可诱导CYP450酶如CYP2E1的上调。UDP葡糖醛酸基转移酶1家族多肽A1(UGT1A1)是目前发现唯一可代谢胆红素的酶,其功能受损会导致胆红素蓄积进而引发肝毒性。据报道[58],NF-κB的激活可抑制UGT1A1的表达,从而加重炎症过程中的高胆红素血症。微管相关蛋白1A/1B-轻链3(LC3)、泛素结合蛋白P62是自噬过程中的关键蛋白。PM可通过影响LC3、P62蛋白的表达,增加肝细胞自噬,导致肝损伤[59]。综上所述,PM可能通过抑制PI3K/Akt、Nrf2信号通路,下调UGT1A1表达,上调CYP2E1表达,影响LC3、P62蛋白表达,诱导DILI发生。
8 小结
PM通过调控JNK、ROS/ATM/P53通路改变线粒体通透性破坏线粒体功能、介导AMPK/FXR、FXR/SHP通路加重胆汁淤积、参与TLR4/MyD88/NF-κB、TLR4/TRIF/IRF-3通路诱导免疫应激、激活PERK/ATF4/CHOP通路诱发ERs、抑制PPARγ、PI3K/Akt、Nrf2/Keap1通路加重炎症反应与氧化应激等多种途径协同作用共同导致肝细胞死亡,多靶点、多途径、多层次诱导DILI的发生发展(图4)。

图4 PM致DILI信号通路Figure 4 Signaling pathways of polygonum multiflorum-induced DILI
PM致肝毒性信号通路仍有待更多研究探索,如高剂量的PM可引起氨基酸、脂质和能量的代谢紊乱,导致三羧酸循环功能障碍,其中涉及哪些信号通路;P53信号通路在调节细胞铁死亡中发挥着重要作用,PM调控的ROS/ATM/P53通路与肝细胞铁死亡有何关系;PM是否通过其他相关通路调控肝细胞死亡等等。深入剖析不同信号通路之间的作用机制,有助于在临床使用PM时通过调节相关通路的激活或抑制降低其肝毒性发生风险,为防治PM致DILI提供潜在作用靶点。
利益冲突声明:本文不存在任何利益冲突。
作者贡献声明:梁子晗负责课题设计,资料分析,撰写论文;李佳辉、程爽、袁卓雅、荣文雅、刘亚杰、郝玉洁参与收集数据,修改论文;王睿林负责拟定写作思路,指导撰写文章并最后定稿。
