ATP结合盒亚家族B成员4(ABCB4)基因突变相关性肝硬化合并胆囊结石1例报告
2024-03-28刘文迪王芃胡和平周华邦
刘文迪, 王芃, 胡和平, 周华邦
海军军医大学东方肝胆外科医院肝胆内科, 上海 200438
ATP结合盒亚家族B成员4(ATP binding cassette subfamily B member 4,ABCB4)基因位于人类第7号染色体长臂2区1带1亚带(7q21.1),编码生成人类的多药耐药蛋白3(multidrug resistance protein 3,MDR3),是一种主要表达于肝细胞的毛细胆管膜上的P糖蛋白,是磷脂输出泵,将磷脂从肝细胞转运到胆管,是磷脂分泌的限速步骤[1-3]。ABCB4基因突变导致MDR3功能损害时,出现磷脂转运障碍,使胆汁中磷脂减少或缺乏,轻者导致胆管损伤、胆石症、妊娠期肝内胆汁淤积症(intrahepatic cholestasis of pregnancy,ICP)等,重者可发展至门静脉高压、肝硬化、晚期肝病,甚至原发性肝脏、胆道恶性肿瘤[4-6]。为提高对该基因突变所致疾病谱的认识,现将笔者团队收治的1例ABCB4基因突变相关肝硬化合并胆囊结石病例报道如下。
1 病例资料
患者男性,15岁,因“纳差半年,发现肝功能异常、胆囊结石半月余”于2021年9月13日入院。患者半年前无诱因出现纳差,伴厌油腻,无皮肤巩膜黄染、畏寒、发热、腹胀、腹痛等不适,当时未予重视,但纳差、厌油腻症状逐渐加重,遂于2021年8月22日至当地县医院查肝功能:TBil 39.4 µmol/L,DBil 17.2 µmol/L,Alb 51 g/L,ALT 289 U/L,AST 187 U/L,ALP 452 U/L,GGT 1 036 U/L;腹部超声:胆囊炎、胆囊结石、脾大,诊断慢性结石性胆囊炎,给予头孢他定抗感染、甘草酸二胺降酶、奥美拉唑护胃等治疗,上述不适稍微好转。2021年8月28日复查肝功能:ALT 171 U/L,AST 92 U/L,GGT 809 U/L。患者为行腹腔镜胆囊切除于2021年9月13日入本院。既往史:平时健康状态良好,否认乙型肝炎、丙型肝炎等传染病史,无高血压、糖尿病等病史。个人史:无特殊。家族史:患者姑姑妊娠时有胆汁淤积病史;祖父、父亲、姑姑均有胆囊结石病史。入院查体:正常面容,全身皮肤巩膜无异常,心肺检查无异常,腹软、无压痛、反跳痛,墨菲征阴性,肝肋下未触及,脾脏左锁骨中线肋下4 cm,质地韧,无压痛。入院查肝功能:TBil 17.6 µmol/L,DBil 12.6 µmol/L,Alb 42.7 g/L,ALT 219 U/L,AST 177 U/L,ALP 443 U/L,GGT 846 U/L,TBA 43.2 µmol/L;血常规:WBC 3.64×109/L,Hb 143 g/L,PLT 105×109/L;凝血功能:正常;尿常规:尿胆原(+),其余未见异常;心电图:窦性心动过缓伴不齐;胸片:心肺未见明显异常;腹部超声:胆囊炎、胆囊结石(胆囊颈部1枚,10.2 mm×3.8 mm)、脾大;磁共振胰胆管成像(MRCP)+磁共振成像(MRI):肝硬化、脾大、肝脾缘少许积液、食管下段胃底静脉曲张、胆囊炎、胆囊结石、肝门部及门腔静脉间隙内淋巴结增大(图1a~c);胃镜:充血渗出性胃炎(胃窦,轻度)、十二指肠球炎、食管下段静脉轻度曲张(图1d);血铜蓝蛋白、血清铜、24 h尿铜未见异常;裂隙灯检查示K-F环阴性;自身免疫性肝病抗体、抗核抗体谱均未见异常。根据患者发病年龄较小,有肝硬化及门静脉高压症表现(脾大、食管下段静脉曲张),起初考虑肝豆状核变性(Wilson病)可能性大,但血清铜蓝蛋白、血清铜、24 h尿铜、K-F环均未见异常,因此,Wilson病诊断证据不足。

图1 肝脏增强MRI及胃镜结果Figure 1 Liver enhanced MRI and gastroscopy
患者发病年龄小,不仅有肝硬化、门静脉高压,还有胆囊结石;肝功能结果显示转氨酶升高,且有明显胆汁淤积表现;此外,其姑姑有ICP。因此,患者转氨酶升高、肝内胆汁淤积、肝硬化、胆囊结石可能由基因突变所致。与患者及其亲属沟通并取得知情同意后,行遗传疾病全外显子突变基因检测,结果显示ABCB4基因7号染色体2处杂合突变(图2),提示此突变可能与进行性家族性肝内胆汁淤积3型(progressive familial intrahepatic cholestasis type 3,PFIC3)、ICP3(妊娠期肝内胆汁淤积症3型)、低磷脂相关胆石症(low phospholipid-associated cholelithiasis syndrome,LPAC)相关(表1)。

表1 患者基因突变信息及可能出现的临床疾病表型Table 1 Genetic mutation information of patients and possible clinical disease phenotypes
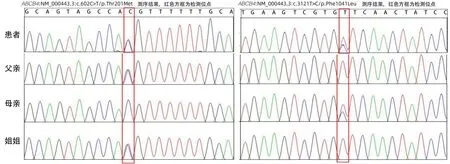
图2 患者及其父母等全外显子基因测序结果Figure 2 Sequence diagram of the whole Exon gene of the patient and his parents
进一步对患者的父母、患者的姐姐验证检查,提示三者均含有1处杂合突变,最后诊断为ABCB4基因突变相关肝硬化合并胆囊结石。治疗上给予熊去氧胆酸胶囊利胆、甘草酸二胺降酶改善肝功能,考虑此病可能影响脂溶性维生素吸收,故进一步检查患者血清脂溶性维生素水平,结果显示,患者25-羟基维生素D、维生素A均降低,分别为16.09 ng/mL(正常值:20.00~80.00 ng/mL)、0.26 mg/L(正常值:≥0.30 mg/L),因此加用复合维生素治疗,患者纳差、厌油腻症状逐渐消失。2023年1月12日,患者复查肝功能基本恢复正常(TBil 17.0 µmol/L、DBil 5.8 µmol/L、Alb 49 g/L、ALT 33 U/L、AST 28 U/L、ALP 82 U/L、GGT 52 U/L)。
2 讨论
ABCB4突变可造成MDR3表达、稳定性或活性改变。MDR3属于ATP结合盒超家族成员,其功能为磷脂酰输出酶,介导肝细胞内的磷脂酰胆碱从磷脂双分子层的内侧转运至膜外的胆汁中[4,7]。在生理状况下,胆汁中磷脂能够与胆盐以适当比例(2∶1~3∶1)形成稳定的混合微粒,将胆盐乳化,并使胆固醇达到最大溶解,从而达到一方面降低胆盐对细胞的毒性去垢作用,保护肝细胞与胆管细胞免于损伤;另一方面避免胆固醇析出,降低结石发生风险。但当ABCB4基因突变导致MDR3缺陷时,胆汁中的磷脂缺乏,肝细胞与胆管细胞由于长时间暴露在胆盐的毒性作用下,产生持续的炎症和纤维性增生,导致胆汁淤积、肝纤维化,甚至肝硬化、门静脉高压、终末期肝病以及恶性肿瘤发生。胆汁中的胆盐也因混合微粒未正常形成或形成速度过慢而不能及时转运,极易析出形成结晶。此外,由于胆固醇的溶解度受胆汁酸和磷脂浓度影响,因此磷脂酰胆碱转出率下降也易导致肝内外胆固醇结晶及胆石症的发生[4,7-9]。
综上所述,ABCB4基因突变导致MDR3缺陷时,根据其临床表型差异,可产生一系列疾病谱,包括PFIC3、LPAC、ICP、成人胆汁纤维化或肝硬化、药物性肝损伤、短暂性新生儿胆汁淤积症[10-18],甚至原发性肝脏、胆道恶性肿瘤[1,19-21]。此外,部分ABCB4基因突变患者在生长、发育过程中可能经历表型重叠。治疗上,熊去氧胆酸具有亲水、肝毒性小特点,以及免疫调节、抗炎和抗凋亡作用,是ABCB4缺乏症患者的一线治疗药物,可用于ABCB4各型突变的患者[15]。对发展至终末期肝病的患者,原位肝移植仍是唯一的治疗方法。
本例患者系青少年,肝功能不仅表现为转氨酶升高,而且胆汁淤积酶谱也明显升高,影像学已有肝硬化、脾大、胆囊结石,胃镜提示食管静脉轻度曲张,在排除病毒性以及基本排除Wilson病和其他自身免疫性肝病等因素后,结合患者发病年龄较小,直系亲属中多人有胆囊结石病史,以及患者姑姑有ICP发作史,考虑该患者肝内胆汁淤积、肝硬化、胆囊结石可能为遗传因素所致;进一步行二代测序明确诊断为ABCB4基因突变相关性胆囊结石、肝硬化。同时,患者还存在脂溶性维生素吸收不良,导致血清脂溶性维生素水平降低。给予熊去氧胆酸利胆、补充复合维生素治疗后,患者乏力、厌油腻症状消失,肝功能基本恢复正常。
综上,在临床工作中对不明原因胆汁淤积、转氨酶升高、肝硬化,尤其是合并胆石症患者,需考虑到ABCB4基因突变相关疾病可能,需要十分详细地询问病史及家族史,在完善实验室、影像学等检查后,仍不能明确病因,可考虑行遗传性疾病突变基因检测以便明确诊断。
伦理学声明:本例报告已获得患者及家属知情同意。
利益冲突声明:本文不存在任何利益冲突。
作者贡献声明:刘文迪、王芃负责收集数据,资料分析,撰写文章;胡和平、周华邦负责课题设计,拟定写作思路,修改文章并最后定稿。
