选择性雌激素受体降解剂研究进展
2024-03-25房文通郭明鑫曹孟妲殷咏梅
房文通,郭明鑫,曹孟妲,殷咏梅
(1.南京医科大学第一附属医院/江苏省人民医院药学部,江苏 南京 210029; 2.宜兴市人民医院药学部,江苏 无锡 214200;3.东南大学附属中大医院药学部,江苏 南京 210009;4.南京医科大学第一附属医院/江苏省人民医院肿瘤科,江苏 南京 210029)
根据世界卫生组织国际癌症研究机构发布的2020年全球癌症数据,乳腺癌已超过肺癌,成为全球发病率第一的癌种[1]。乳腺癌患者中约70%为雌激素受体(estrogen receptor,ER)和(或)孕激素受体(progesterone receptor,PR)阳性,称为激素受体(hormone receptor,HR)阳性乳腺癌。内分泌治疗是HR阳性乳腺癌治疗的基石[2],但部分患者会因内分泌治疗耐药出现肿瘤复发、进展。耐药通常由多种机制驱动,ERα编码基因ESR1突变是常见的原因之一。如何解决ESR1突变引起的耐药问题,保证HR阳性乳腺癌内分泌治疗的持续有效是一个重要的临床问题。选择性雌激素受体降解剂(selective estrogen receptor down-regulators or degraders,SERD)氟维司群对ESR1突变患者仍有一定疗效[3]。然而氟维司群药代动力学特性差,生物利用度低,只能肌肉给药,达稳态浓度时间长,靶点暴露量低等[3],限制了其临床应用。因此,迫切需要改进SERD,改善其药动学性质,提高其药效学性能。本文对乳腺癌治疗药物SERD的研发和临床应用进展进行综述,以期为乳腺癌的药物治疗提供参考。
1 雌激素受体阳性乳腺癌内分泌治疗的现状
雌激素与ER结合后,可激活下游的信号通路,促进肿瘤细胞生长与增殖。通过抑制或阻断雌激素对肿瘤细胞的刺激作用治疗ER阳性乳腺癌的方式称为内分泌治疗[2]。目前,ER阳性乳腺癌内分泌治疗药物分3类:雌激素受体调节剂(selective estrogen receptor modulator,SERM)、芳香化酶抑制剂(aromatase inhibitor,AI)和SERD,其具体作用机制如图1所示。SERM代表药物为他莫昔芬(tamoxifen),其结构与雌激素相似,在靶器官内与雌激素竞争结合ER,阻断雌激素与ER的结合。他莫昔芬在不同组织中对ER的作用不同,在乳腺组织中是ER拮抗剂,但在子宫中则是一种ER弱激动剂,会增加子宫内膜癌的风险。绝经后女性的雌激素主要来源于雄激素转化,芳香化酶是催化体内雄激素向雌激素转化的关键酶和限速酶。AI能特异性导致芳香化酶失活,阻断芳构化反应,抑制雌激素生成。非甾体类AI药物(如阿那曲唑和来曲唑)对芳香化酶的抑制是可逆性的,而甾体类AI(如依西美坦)对芳香化酶的抑制是不可逆的。AI的整体耐受性良好,但潮热、盗汗、关节/肌肉疼痛和骨质疏松症等副作用,也会影响其依从性[4]。首个SERD类药物是氟维司群,于2002年上市。氟维司群与ERα的结合引起ERα结构变化,导致其表面疏水性增加,吸引E3泛素连接酶和蛋白酶体,导致ERα降解,使雌激素失去作用靶点。氟维司群不引起子宫内膜癌和血栓栓塞等不良反应[5],目前已批准用于乳腺癌的新辅助、一线和二线治疗。
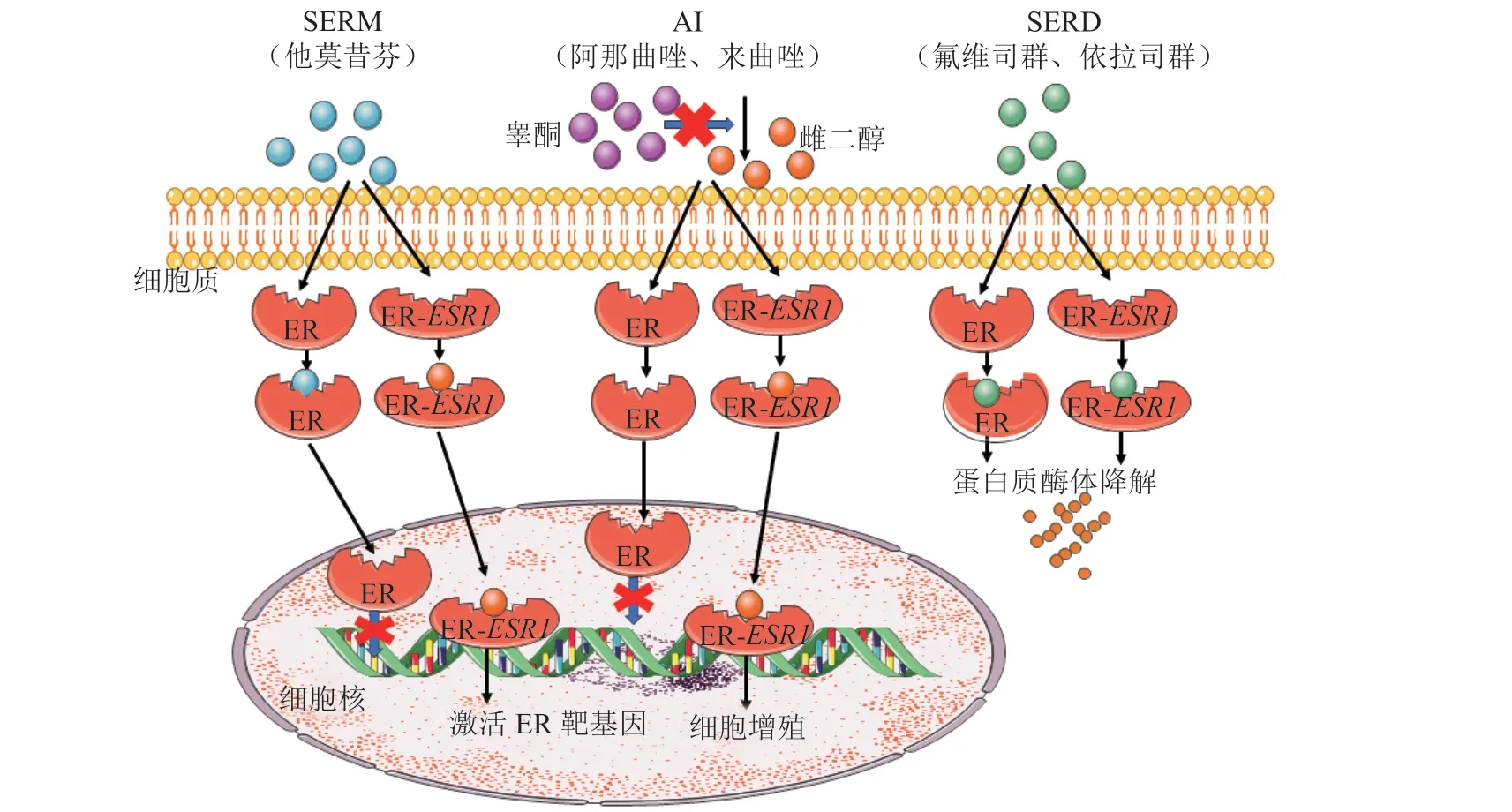
图1 乳腺癌内分泌治疗药物的作用机制Figure 1 Mechanism of endocrine therapy drugs in breast cancer
晚期乳腺癌患者内分泌治疗常存在耐药,耐药通常由多种机制驱动,ERα编码基因ESR1突变是常见的原因之一。晚期乳腺癌患者AI治疗后,约20% ~ 40%患者出现ESR1突变,其发生率因转移灶位置而异[6]。ESR1突变多位于配体结合结构域,最常见的突变类型是D538G和Y537S。ESR1配体结合域突变后,无需雌激素也可激活ER及其信号通路[6]。在临床前研究中,氟维司群对ESR1突变体较野生型敏感性显著下降。然而,临床研究中氟维司群对ESR1野生型及突变型晚期乳腺癌患者具有相同的疗效[3]。由于存在多种ESR1突变以及个体差异,氟维司群对不同ESR1突变型乳腺癌患者的疗效有所不同。因此,针对ESR1突变型乳腺癌的最佳治疗策略需综合患者具体情况制定个性化治疗选择。此外,考虑到氟维司群药代动力学性质较差,具有生物利用度低,只能肌肉给药,达稳态浓度时间长,靶点暴露量低等缺点[3],因此迫切需要改进SERD以获得更好的药动学和药效学数据。研究人员一方面探索药动学性质更好的SERD,可口服的elacestrant、camizestrant、giredestrant等[7]应运而生,另一方面探索降解ER的新技术,如蛋白水解靶向嵌合体(proteolysis-targeting chimera,PROTAC)[8-9]、分子胶降解(molecular glue degrader,MGD)[10]等,均取得了许多新进展。
2 口服选择性雌激素受体降解剂的开发
2.1 结构特点与开发历程
开发口服SERD的主要障碍是生物利用度低和不可耐受的毒性。新型SERD呈非甾体结构,含丙烯酸侧链或碱性侧链,可引起ERα构象变化,导致受体疏水区暴露,成为蛋白降解的靶点。含丙烯酸侧链的口服SERD是GW5638(1,他莫昔芬的类似物)的活性代谢产物,如GW7604(2)、AZD9496(3)、GDC0810(4)、G1T48(5)、LSZ102(6)和ZN-c5(7),这些药物不仅在内分泌治疗敏感和ESR1突变的临床前模型中均显示出抗肿瘤活性,与细胞周期蛋白依赖性激酶(cyclin-dependent kinase,CDK)4/6抑制剂联合使用也显示协同效应[11]。然而在Ⅰ期临床研究中,大多数含丙烯酸侧链的SERD疗效有限,耐受性较差,因此停止了开发[11-12]。
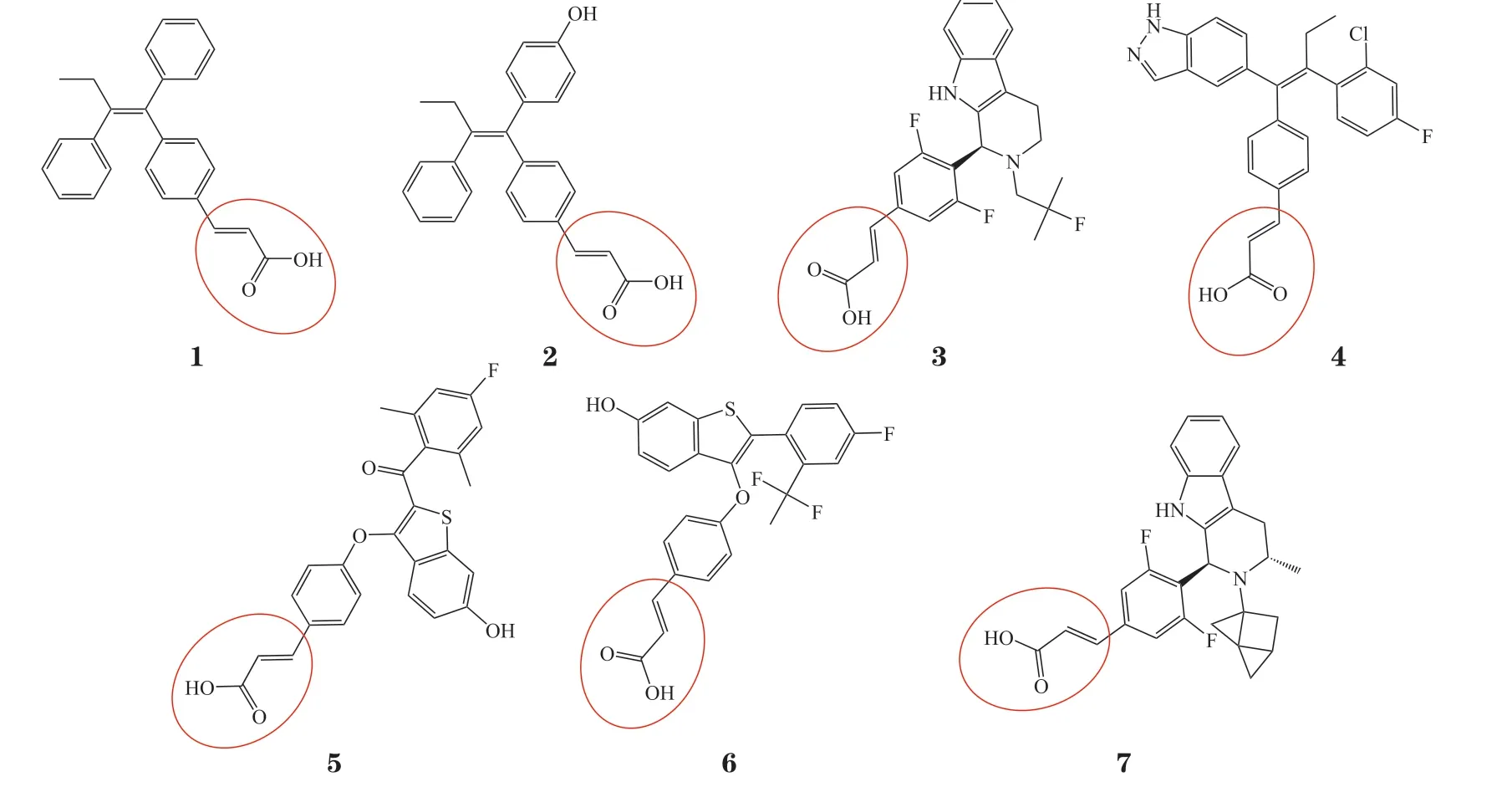
含碱性侧链的口服SERD,如elacestrant(8)、camizestrant(9)、giredestrant(10)等,不仅生物利用度高,且对内分泌治疗敏感和ESR1突变的患者均有效[4,13]。目前已有多项含碱性侧链的口服SERD临床研究的数据公布,研究最为顺利的elacestrant(通用名:依拉司群,商品名:Orserdu)已于2023年上市[7],camizestrant和imlunestrant(11)的Ⅱ期临床研究取得阳性结果,顺利进入Ⅲ期临床研究[4]。Amcenestrant(12,SAR439859)的临床研究并不顺利,其单药治疗的Ⅱ期临床研究(AMEERA-3和AMEERA-4)[14-15]和联合哌柏西利治疗的Ⅲ期临床研究(AMEERA-5)[16]均失败,目前已终止临床开发。Giredestrant对比医生选择的内分泌治疗方案(physician choice of endocrine monotherapy,PCET)在既往经治ER阳性/HER2阴性局部晚期或转移性乳腺癌患者中疗效和安全性的Ⅱ期临床研究(acelERA)[17]也失败了,但仍在推进Ⅲ期临床研究。
2.2 已上市的药物
2023年1月27日,美国食品和药品管理局(Food and Drug Administration,FDA)批准Menarini子公司Stemline Therapeutics的依拉司群(elacestrant)用于既往至少接受过一线内分泌治疗后进展的ER阳性/HER2阴性、ESR1突变的绝经后女性或成年男性晚期或转移性乳腺癌患者,这也是FDA批准的首个口服SERD。Elacestrant的推荐剂量为345 mg,与食物同服,每日1次,直至疾病进展或不可耐受的毒性。FDA还批准了guardant360 CDx检测作为辅助诊断,用于识别接受elacestrant治疗的乳腺癌患者[7]。

2.2.1 药动学Elacestrant在每日43 ~ 862 mg(推荐剂量的0.125倍 ~ 2.5倍)的剂量范围内,峰浓度(maximum concentration,Cmax)和浓度-时间曲线下面积(area under the concentration-time curve,AUC)值与剂量呈正比。Elacestrant的口服生物利用度约为10%,达峰时间为1 ~ 4 h。Elacestrant口服后第6天达到稳态血药浓度,其平均累计率是AUC0-24h的2倍。99%以上的elacestrant与血浆蛋白结合,与浓度无关。Elacestrant主要经细胞色素P450酶(cytochrome p450,CYP)3A4亚型代谢,其次由CYP2A6和CYP2C9代谢。单次口服345 mg放射性标记的elacestrant后,82%(34%原型)从粪便中排泄,7.5%(<1%原型)从尿中排出。Elacestrant的消除半衰期为30 ~ 50 h[7,18-19]。由于elacestrant是CYP3A4底物,同时使用强效或中度CYP3A4抑制剂会增加elacestrant的暴露(可能增加elacestrant不良反应),而同时使用强效或中度CYP3A4诱导剂会减少elacestrant的暴露(可能降低elacestrant疗效)。因此,应避免elacestrant与强效或中度CYP3A4抑制剂/诱导剂合用[7,18-19]。
Elacestrant是P-糖蛋白(P-glycoprotein,P-gp)和乳腺癌耐药蛋白(breast cancer resistance protein,BRCP)的抑制剂,与P-gp或BRCP底物合用会增加这些底物的浓度,可能增加这些底物相关不良反应的风险[7,18-19]。
2.2.2 药效学EMERALD是一项多中心、随机、开放标签、阳性对照、Ⅲ期临床试验,入组既往接受过一线或二线内分泌治疗(包含CDK4/6抑制剂)、不超过1次化疗的ER阳性/HER2阴性晚期或转移性绝经后乳腺癌患者,允许患者既往接受过氟维司群治疗。该研究共纳入477例患者,随机分为elacestrant组(n= 239)和PCET组(n= 238)[20],中位随访时间为15.1个月。该研究结果显示,在总体人群中,elacestrant组较PCET组的中位无进展生存期(progression free survival,PFS)适度改善(2.8个月vs1.9个月,HR = 0.70,95%CI:0.55 ~ 0.88,P= 0.001 8),elacestrant组疾病进展或死亡风险降低30%,12个月无进展生存率有所提高(22.3%vs9.4%)。在228例(48%)ESR1突变患者中,elacestrant组较PCET组中位PFS显著改善(3.8 个月vs1.9个月,HR = 0.55,95%CI:0.39 ~ 0.77,P= 0.000 5)[7,19,21]。
2.2.3 不良反应在Ⅰ期临床研究[22]和EMERALD研究[20]中,elacestrant单药治疗与其他内分泌治疗方案均具有可控的安全性。Elacestrant最常见的不良反应(发生率≥10%)为肌肉骨骼疼痛、肝功能异常、消化道功能异常、血脂代谢紊乱、血红蛋白降低、低钠、肌酐升高、头痛和潮热等[7,23]。
2.2.4 特殊人群用药年龄(24 ~ 89岁)、体质量(41 ~143 kg)及性别对elacestrant的药动学未见显著影响。轻度肝功能不全患者(child-pugh A)不需要调整elacestrant剂量。中度肝功能不全患者(childpugh B)[7]elacestrant的AUC增加了83%,elacestrant剂量需降低至258 mg/日。尚无在严重肝功能不全患者(child-pugh C)及妊娠期或哺乳期女性中elacestrant应用的数据[7,18]。
2.3 处于Ⅲ期临床研究的药物
2.3.1 camizestrantCamizestrant(AZD9833)已在Ⅰ期和Ⅱ期临床研究中取得了阳性结果。Ⅰ期临床试验SERENA-1(NCT03616587)纳入已接受过一线或二线内分泌治疗及化疗的ER阳性/HER2阴性晚期乳腺癌患者,camizestrant单药或联合CDK4/6抑制剂均显示出一定的抗肿瘤活性[24]。Ⅱ期临床试验SERENA-2[25]在不超过一线内分泌治疗后复发或进展的晚期HR阳性/HER2阴性绝经后女性乳腺癌中比较camizestrant(75和150 mg)与氟维司群的疗效。Camizestrant 75 mg剂量组、150 mg剂量组与氟维司群组中位PFS分别为7.2个月(HR = 0.58,95%CI:0.41 ~0.81)、7.7个月(HR = 0.67,95%CI:0.48 ~ 0.92)和3.7个月。Camizestrant两个剂量水平在PFS方面均获益[26-27]。
目前多个camizestrant的Ⅲ期临床试验正在进行。SERENA-4试验(NCT04711252)研究camizestrant联合哌柏西利在ER阳性/HER2阴性晚期乳腺癌一线治疗中的作用。SERENA-6试验评估camizestrant联合CDK4/6抑制剂一线治疗HR阳性、ESR1突变转移性乳腺癌患者的疗效和安全性。同时,CAMBRIA-1[28]和CAMBRIA-2研究将探索camizestrant在早期乳腺癌中的作用。
2.3.2 giredestrantⅠ期临床研究发现,giredestrant(GDC-9545)每日10、30、90/100、250 mg均有效,且耐受性良好[29]。Ⅱ期临床研究(acelERA研究)[17]纳入既往经治ER阳性/HER2阴性局部晚期或转移性乳腺癌患者,比较giredestrant与PCET的疗效和安全性,中位随访7.89个月,两组中位PFS(5.6个月vs5.4个月,HR = 0.81,P= 0.18)未达到统计学显著差异,但giredestrant组的临床获益率(clinical benefit rate,CBR,31.8%vs21.1%)和客观缓解率(objective remission rate,ORR,12.6%vs7.2%)更高。Giredestrant在ESR1突变患者中PFS获益更加显著(5.3个月vs3.5个月,P= 0.061)[30],提示giredestrant可能在ESR1突变患者中具有较好应用前景。
2023年欧洲肿瘤内科学(european society for medical oncology,ESMO)会议公布的Ⅰ/Ⅱ期MORPHEUS BC研究结果,旨在评估giredestrant联合治疗ER阳性/HER2阴性局部晚期/转移性乳腺癌的安全性和有效性。截至2023年5月31日,giredestrant单药组的ORR为0%,而联合组为20.8%,中位PFS分别为4.67和5.37个月;联合方案治疗时,蛋白激酶B通路改变和未改变患者的ORR分别为30.0%和15.4%,中位PFS分别为7.47和2.96个月[31]。评估giredestrant或来曲唑联合哌柏西利对ER阳性/HER2阴性局部晚期或转移性乳腺癌患者的疗效和安全性的Ⅲ期临床研究(pionERA)正在进行中。
2.3.3 imlunestrantImlunestrant(LY3484356)的Ⅰa/b期临床研究(EMBER)在ER阳性/HER2阴性晚期乳腺癌中评估imlunestrant±依维莫司/阿培利司的可行性。截至2023年3月9日,在CDK4/6抑制剂经治进展后二线治疗的人群中,imlunestrant单药、imlunestrant+依维莫司及imlunestrant+阿培利司的中位PFS分别为6.5、15.9和9.2个月,ORR分别为8%、21%和58%,CBR分别为42%、62%和62%,最常见不良反应为恶心和腹泻[32-33]。进一步的EMBER-2研究也证实imlunestrant(400或800 mg)对Ⅰ~Ⅲ期ER阳性/HER2阴性绝经后女性乳腺癌患者的疗效[34]。目前,imlunestrant正在进行Ⅲ期临床研究,EMBER-3和EMBER-4研究旨在评估imlunestrant单药或联合治疗在ER阳性/HER2阴性局部晚期或转移性乳腺癌中的疗效。
2.3.4 brilanestrantBrilanestrant(13,GDC-0810)在Ⅰa/Ⅰb/Ⅱa期剂量爬坡试验中展现了良好的安全性和耐受性。总体人群和携带ESR1突变的患者中,部分缓解(总体:4%;ESR1突变:4%),病情稳定(总体:39%;ESR1突变:42%),非完全缓解/非进展性疾病(总体:13%;ESR1突变:12%)均有获益。在剂量递增(n= 16,39%)和剂量扩增试验(n= 24,22%)中也观察到临床获益(持续时间≥24周)[35]。中国国家药品监督管理局药品审评中心已批准brilanestrant单药作为二线疗法的Ⅲ期临床试验。

2.4 处于Ⅰ/Ⅱ期临床研究的药物
2023年ESMO大会介绍了正在进行Ⅰ/Ⅱ期研究的一些口服SERD新药,如palazestrant(OP-1250)、vepdegestrant和LX-039(14)等。这些新药研究将进一步推动口服SERD在HR阳性晚期乳腺癌中的应用。
OP-1250是一种兼具SERD和ER拮抗活性的口服小分子药物。Ⅰ/Ⅱ期剂量递增和剂量扩展研究已确定120 mg每日1次为最大耐受剂量;截至2023年3月17日,86例接受该剂量的患者,CBR为40%,中位PFS为4.6个月,ESR1突变亚组的中位PFS为5.6个月;在二线或三线治疗时,CBR为48%,中位PFS为7.2个月,ESR1突变亚组的中位PFS为7.3个月[36-37]。
LX-039的首次临床试验结果在2023年ESMO大会中公布,共纳入44例患者,CBR为40.0%,中位PFS为5.5个月,中位OS尚未达到[38]。此外,GDC0927(15)、FWD1802、D0502(taragarestrant)、HS234等药物也处于临床研究阶段。
3 基于新型雌激素受体降解技术开发的药物
具有碱性侧链的口服SERD用药方便,对内分泌治疗敏感和ESR1突变的乳腺癌患者均有效。然而口服SERD需较高浓度“占据”靶点ER足够长时间才能发挥作用,这也是小分子药物的共性同缺点。因此研究人员还在探索降解ER的新技术,只需与靶蛋白有一定的结合率即可降解靶蛋白,可作用于“不可成药”或突变的靶点。PROTAC[8-9]、MGD[10]等新型技术的应用使SERD开发取得了重大突破(见图2),但基于这些技术开发的药物大多数处于临床前研究阶段。
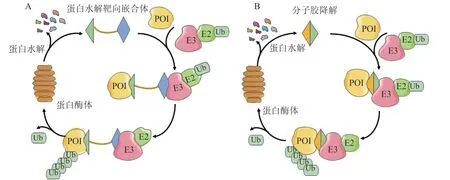
图2 蛋白水解靶向嵌合体(A)和分子胶降解(B)技术作用机制的示意图Figure 2 Schematic diagram of the mechanism of proteolysis-targeting chimera(A) and molecular glue degrader(B)
3.1 蛋白水解靶向嵌合体
PROTAC由3个关键结构组成:一侧是靶蛋白配体,另一侧是E3泛素连接酶配体,中间是连接这2个配体的连接子。PROTAC是一个双功能小分子,一个配体负责结合靶蛋白,另一个配体负责结合E3泛素连接酶。当PROTAC两端同时结合上靶蛋白和E3泛素连接酶时,靶蛋白被加上泛素化标签,进而被泛素-蛋白酶体系降解,而剩下的PROTAC则能够多次循环发挥作用[39]。不同于传统小分子药物和目标蛋白“一对一”的作用关系,PROTAC和靶蛋白以“一对多”的方式发挥作用。目前已开发出包括ER靶向型PROTAC在内的50多种PROTAC[8]。
AVR-471是一种应用PROTAC技术的新型ER降解剂,其一端可结合ER,另一端可结合E3泛素连接酶CRBN。Ⅰ期临床试验表明,AVR-471的ER降解率为89%,在34例患者中的CBR为41%。AVR-471具有良好的耐受性,最常见的治疗相关不良反应为恶心(24%)、疲劳(12%)和呕吐(10%),主要为1级,2例3级不良反应,其中1例因3级血栓栓塞而停用ARV-471[4,9]。ARV-471在转移性乳腺癌中的Ⅰ期剂量递增研究、与哌柏西利联用的Ⅰb/Ⅱ期单药剂量扩增试验[4]、经治(VERITAC-2,NCT05654623)或未经治(VERITAC-3,NCT05909397)晚期乳腺癌患者中的Ⅲ期临床研究也相继开展。
目前国内也开发了一些ER PROTAC药物,如HRS-1358已进入临床研究阶段,还有一些ER PROTAC药物已申请专利。PROTAC药物的缺点是相对分子质量较大,细胞渗透性和药动学性质较差。此外,PROTAC药物也需要靶蛋白具有可链接的结合口袋,这也限制了其应用。
3.2 分子胶降解
分子胶是指一类能使2个以上的蛋白质之间以糊状形式相连接的小分子。MGD是指分子胶诱导或稳定泛素连接酶和靶蛋白之间的蛋白-蛋白相互作用,导致蛋白质泛素化和随后的蛋白酶体降解,分子胶随后解离[4,10]。虽然MGD与PROTAC均为泛素化降解,但MGD具有独特的作用特点:1)分子胶可以结合没有结合口袋的靶蛋白(非可成药性靶标),诱导或加强靶蛋白与E3泛素连接酶之间的相互作用[10];2)分子胶化学结构简单,相对分子质量小,细胞通透性高,口服吸收好,具有良好的成药性,为靶向蛋白降解领域的药物开发提供了新的研究思路;3)MGD可以在胞浆中降解新生的ER,并降解ESR1及其他参与内分泌治疗抵抗的蛋白,如磷脂酰肌醇-3-激酶、CDK4/6等。临床前和早期临床数据表明,基于MGD开发的药物可能比SERD和ER PROTAC具有更高的效力,且毒性更小[4,40]。
3.3 其他
溶酶体也是蛋白降解的重要场所,靶向溶酶体体系蛋白降解的新技术也发展迅速,如溶酶体靶向嵌合体、自噬靶向嵌合体和自噬小体绑定化合物等[41-42]。这些技术介导的蛋白质降解,类似PROTAC,通过将靶标蛋白泛素化从而导致降解的发生,但不同的是,前者是通过溶酶体的方式而PROTAC是通过蛋白酶体的方式实现的降解。
靶蛋白降解技术,不但可靶向“不可成药”或耐药靶点,而且实现了传统小分子难以实现的选择性和高活性。虽然目前仅有ARV-471进入临床研究,相信以后会有更多该类新型药物进入临床研究,并改善HR阳性乳腺癌的预后。同时,作为一种新技术,仍存在潜在的安全隐患,一些副作用、毒性或免疫反应等问题可能需要更深入的研究和评估,确保该技术的安全性和可行性。
4 结语
内分泌治疗是HR阳性乳腺癌最重要的治疗方法。SERD的开发是近年来内分泌治疗研究的重要进展[43]。首个口服SERD药物elacestrant已于2023年上市,camizestrant、giredestrant、amcenestrant、imlunestrant等药物也进入Ⅲ期临床阶段,还有部分药物处于Ⅰ/Ⅱ期临床阶段。口服SERD可有效地靶向ER野生型和ESR1突变型乳腺癌,促进ER降解并破坏下游信号传导,表现出良好的有效性和安全性。虽然也有口服SERD未达到试验终点,可能与缺乏ESR1突变肿瘤的富集有关,因此强烈推荐正在进行的口服SERD试验中检测ESR1突变。此外,新技术如PROTAC、MGD等在降解ER方面也发挥了重要作用,ER PROTAC新药AVR-471已进入临床研究并初步证实了其安全性和有效性。目前正在积极探索这些药物在乳腺癌内分泌治疗中的潜力,希望能够改善患者的预后。
