晚期激素受体阳性乳腺癌内分泌耐药与靶向治疗药物研究进展
2024-03-25李泽颖杨凡黄香殷咏梅
李泽颖,杨凡,黄香,殷咏梅
(南京医科大学第一附属医院/江苏省人民医院肿瘤科,江苏 南京 210029)
2020年,世界卫生组织发布乳腺癌发病率超过肺癌,成为全球第一大癌症。近年来,虽然癌症整体死亡率有所下降,但2023年美国仍预计有1 958 310例新发癌症病例和609 820例癌症死亡病例[1]。我国每年乳腺癌的发病例数位居全国高发恶性肿瘤第5位,且位居女性恶性肿瘤发病率的首位,对社会与家庭造成了极大的经济负担。
乳腺癌根据雌激素受体(estrogen receptor,ER)和孕激素受体(progesterone receptor,PR)状态、人表皮生长因子受体2(human epidermal growth factor receptor 2,HER2)是否突变以及细胞分子状态可以分为不同的亚型,分别是Luminal A(HER2阴性、ER阳性、PR阳性且高表达、Ki-67低表达)、Luminal B(HER2阴性、ER阳性、PR低表达或阴性、Ki-67高表达)、HER2阳性/激素受体(hormone receptor,HR)阴性(HER2阳性、ER阴性、PR阴性、Ki-67任何状态)、HER2阳性/HR阳性(HER2阳性、ER阳性、PR任何状态、Ki-67任何状态)以及三阴性(ER、PR以及HER2均为阴性)。其中,HR阳性的患者基数最大,约占所有乳腺癌患者的70%。ER和PR是参与乳腺癌生长途径最重要的两类HR,ERα是一种类固醇激素受体和转录因子,可以通过上调类固醇孕激素受体PR,从而与雌激素结合,调节乳腺癌细胞的发生发展[2]。自20世纪70年代以来,ER拮抗剂他莫昔芬(tamoxifen,TAM)一直是乳腺癌内分泌治疗的标准药物。TAM可用于治疗早期、局部晚期和转移性ER阳性乳腺癌,大大降低了绝经前乳腺癌患者复发的风险。由于芳香化酶可催化体内雄激素转变为雌激素,随后,以来曲唑、阿那曲唑和依西美坦为代表的一系列芳香化酶抑制剂(aromatase inhibitor,AI)开始应用于内分泌治疗,它们通过降低雌激素水平,抑制乳腺癌的发展,目前主要用于治疗绝经后的乳腺癌患者。选择性降解ER药物氟维司群与HR结合的亲和力极高,与TAM相比高出约100倍,2002年美国食品和药品管理局(food and drug administration,FDA)批准了氟维司群治疗HR阳性转移性乳腺癌,开启了内分泌治疗的新篇章[3]。此外,还可以通过手术去势或使用卵巢功能抑制剂如戈舍瑞林来降低体内雌激素水平[4]。
虽然内分泌治疗可以使大多数晚期HR阳性乳腺癌患者获益,但有20%的患者可能会出现原发耐药,另有30% ~ 40%患者可能因为继发耐药而病情进展[5]。随着分子生物学和肿瘤生态学研究的不断进步,细胞周期蛋白依赖性激酶4/6(cyclin-dependent kinases 4/6,CDK4/6)抑制剂的问世改变了内分泌治疗格局,大量的生物标志物和治疗靶点逐渐被挖掘,乳腺癌的精准治疗与分层治疗逐步深入研究。内分泌治疗联合分子靶向逐渐成为晚期HR阳性乳腺癌患者的首选方案。本文探讨了晚期HR阳性乳腺癌内分泌治疗的耐药机制及近年来内分泌分子靶向药物的临床研究进展,对于HR阳性乳腺癌的未来治疗策略提出了新的展望。
1 激素受体阳性乳腺癌内分泌治疗的耐药机制
1.1 雌激素受体与芳香化酶编码基因突变
雌激素受体α基因1(estrogen receptor α1,ESR1)可编码转移性乳腺癌患者ERα表达。早在1997年就已发现乳腺癌患者存在ESR1点突变,包括S47T、K531E、Y537N等[6]。近年来,研究人员发现,ESR1配体结合域(ligand binding domain,LBD)的获得性突变是ER阳性转移性乳腺癌耐药的常见驱动因素[7]。在长期使用TAM或AI治疗后,约20%的患者出现LBD结构的突变[8]。最常见的2个突变氨基酸分别是Y537和D538,它们均位于LBD中H12螺旋的C端。H12是激活功能2结构域(activation function 2 domain,AF2)的关键成分,决定受体的激动或拮抗状态。Y537S突变和较小程度的D538G突变可使H12稳定在激动构象中,减轻其对TAM的亲和力,从而促进对拮抗剂的抗性[9]。此外,ESR1突变型乳腺癌细胞可表现为增殖样和干细胞性表型,其有丝分裂信号增强,有更强的上皮-间质转化和转移倾向[10]。Gelsomino等[11]研究发现,过表达ESR1突变的细胞系可通过ER/缺口受体4(neurogenic locus notch homolog 4,NOTCH4)相互作用,在CD44+/CD24-细胞中富集,表明ESR1突变的乳腺癌细胞干性增强。目前,选择性雌激素受体共价拮抗剂(selective estrogen receptor covalent antagonist,SERCA)代表化合物H3B-5942处于Ⅱ期临床试验阶段,其共价结合位点是半胱氨酸530(C530),该位点在与H3B-5942结合后,无论是否存在Y537S或D538G ESR1突变,均表现为拮抗剂构象;因此,H3B-5942具有优于TAM和氟维司群的抗肿瘤活性,以及优于氟维司群的最大降解能力[12]。此外,有研究表明,ESR1的染色体易位促进了嵌合转录因子的形成,从而驱动了内分泌治疗抵抗[13]。研究人员在复发性乳腺癌细胞中还发现了ESR1基因扩增,接受辅助AI治疗的HR阳性乳腺癌患者中,ESR1扩增可以被特异性选择,从而诱导ESR1相关通路再激活,促进了HR阳性乳腺癌的耐药和复发[14]。在一项纳入394例绝经后接受辅助内分泌治疗患者的临床研究中,有187例存在ESR1基因扩增,提示ESR1扩增可作为绝经后HR阳性早期乳腺癌长期临床结局的独立预测因子[15]。
芳香化酶编码基因CYP19A1可催化睾丸酮、雄烯二酮转化为雌酮和雌二醇。有研究发现,21.5%经AI治疗的复发性乳腺癌患者出现了CYP19A1基因扩增[16]。AI治疗下的乳腺癌细胞缓慢进化,CYP19A1扩增驱动了内源性表观遗传胆固醇生物合成或肿瘤微环境的循环,导致了芳香化酶活性增强,促进了雌激素独立转录,但CYP19A1扩增的乳腺癌对氟维司群和依西美坦等药物仍然敏感,靶向CYP19A1的雌激素受体下调剂(selective estrogen receptor down-regulator,SERD)可能是未来乳腺癌新药的研发方向[16]。
1.2 分子通路异常活化
1.2.1 PI3K/AKT/mTOR信号通路活化的磷脂酰肌醇3激酶(phosphatidylinositol-3-kinase,PI3K)可催化4,5-二磷酸化磷脂酰肌醇(phosphatidyl-inositol 4,5-bisphosphate,PIP2)磷酸化生成3,4,5-三磷酸化磷脂酰肌醇(phosphatidyl-inositol-3,4,5-trisphosphate,PIP3),PIP3通过招募丝氨酸/苏氨酸激酶(serinethreonine kinase,AKT)并引起其构象改变,从而进一步激活下游靶基因,参与调控细胞的增殖和分化。10号染色体上缺失的磷酸化和张力蛋白(phosphate and tension homology deleted on chromosome ten,PTEN)是参与PIP3去磷酸化的主要负调控因子。哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)包括mTOR1和mTOR2,后者可磷酸化AKT,加强其磷酸化激酶的活性,前者则在AKT的下游影响细胞生长和蛋白合成[17]。PI3K/AKT/mTOR通路在细胞代谢、生长、增殖、凋亡和血管生成等细胞活动中发挥重要作用。研究发现,约有30% ~ 40%的HR阳性乳腺癌存在PI3KCA基因突变,加入PI3K抑制剂可明显改善HR阳性患者的预后;此外,ATK和PTEN突变也与内分泌治疗的耐药性相关[18]。最新研究发现,PTEN的缺失激活了PI3Kβ的表达,一方面可以通过作用于AKT激酶促进肿瘤细胞的生长,另一方面则激活了信号转导与转录激活因子(signal transducer and activator of transcription,STAT)介导的免疫抑制;当PI3Kβ或STAT3表达下调时,可以促进抗原呈递和免疫细胞的募集,PI3Kβ抑制剂与抗程序性细胞死亡蛋白-1(programmed cell death protein-1,PD-1)免疫治疗联合可能增强抗肿瘤活性[19]。
1.2.2 酪氨酸激酶信号通路酪氨酸激酶(receptor tyrosine kinase,RTK)是一类在细胞膜上表达的受体,可以启动有丝分裂信号级联。配体-受体结合后诱导同源或异源二聚化和自磷酸化,通过促分裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)、PI3K/AKT和JAK/STAT通路激活下游信号转导。成纤维细胞生长因子受体(fibroblast growth factor receptors,FGFRs)家族包含4个跨膜受体(FGFR1、FGFR2、FGFR3和FGFR4)和一个膜相关受体(FGFR5)。配体与膜受体的结合促进受体二聚化和C端酪氨酸的磷酸化,从而作用于成纤维细胞生长因子受体底物α。有研究表明,FGFR1和FGFR2还可以定位于乳腺癌细胞核,调节基因转录,FGFRs的表达上调,基因的扩增、突变和融合均可诱导乳腺癌的内分泌治疗抗性,靶向FGFR的酪氨酸激酶抑制剂在FGFR融合和突变的实体肿瘤中具有临床疗效;20% ~ 30%的ER阳性乳腺癌患者中HER2表达扩增,ER阳性/HER2阴性转移性乳腺癌细胞可以在内分泌治疗中获得HER2突变,HER2通过与ER、PR相关分子通路发生信号串扰,从而使其对内分泌治疗产生耐药性[20]。Ⅲ期临床试验探索了拉帕替尼联合内分泌治疗作为ER阳性乳腺癌晚期一线治疗方案的可行性,虽然总体上没有改善HER2阴性患者的无进展生存期(progression-free survival,PFS),但仍有一部分HER2阴性患者对拉帕替尼联合内分泌治疗有反应[21-22]。
1.2.3 MAPK信号通路MAPK分子通路中有1型神经纤维瘤病基因(neurofibromatosis type 1,NF1)、鼠类肉瘤病毒癌基因(kirsten rat sarcoma viral,KRAS)、鼠类肉瘤滤过性毒菌致癌同源体b1(v-Raf murine sarcoma viral oncogene homolog B,BRAF)等。有研究对485例ER阳性的浸润性导管癌患者组织进行了分析,结果发现在转移性浸润性导管癌中NF1突变特异性富集,且NF1突变主要见于获得性耐药,通常表现为杂合性缺失,且检测出的循环肿瘤DNA为多克隆,ER阳性乳腺癌体外模型验证了在TAM存在下,NF1表达下调的乳腺癌细胞更具有生长优势[23]。Razavi等[24]分析了1 501例HR阳性乳腺肿瘤基因组测序和相应的临床信息,发现了MAPK通路相关基因如KRAS和叉头框架蛋白A1(forkhead-box protein A1,FOXA1)的表达富集,且与ESR1中的热点突变互斥,MAPK通路中信号分子表达改变的患者对激素治疗反应比较差,这表明MAPK信号通路改变的患者对ER表达水平的依赖性减弱。
1.2.4 细胞周期异常激活正常的细胞周期受到调节因子的严格调控,有丝分裂信号触发后,细胞周期蛋白可以结合并激活细胞周期蛋白依赖性激酶(cyclin-dependent kinases,CDK)。然而在肿瘤细胞中,有丝分裂信号过多或抑制细胞分裂的检查点失活导致了细胞的无序增殖[25]。雌激素可以通过上调细胞周期调节蛋白(包括Cyclin D1和Cyclin E)的转录或通过激活Cyclin依赖性激酶(CDK2和CDK4),导致下游视网膜母细胞瘤基因(retinoblastoma gene,RB1)的过度磷酸化,诱导细胞周期从G1期进入S期。内分泌治疗可减少雌激素依赖性CDKCyclin络合,并在G0、G1期阻断细胞周期进程[26]。在G1期到S期的转变中,雌激素作用的关键效应物有Cyclin D1,内分泌耐药肿瘤细胞可表现为细胞周期调节因子的改变,如Cyclin E、Cyclin D活化,以规避内分泌治疗对G1期向S期转换的抑制作用。此外,Cyclin D1基因的过表达和扩增可导致CDK4/6活性频繁失调,赋予HR阳性乳腺癌不依赖ER的增殖能力,从而削弱AI疗效[27](见图1)。
1.3 表观遗传机制
ERα依赖与共调节蛋白的相互作用来调节自身活性,突出的表观遗传ERα共激活因子包括P160家族、P300/CBP等,其中,P160家族中的类固醇受体共激活因子(steroid receptor coactivator,SRC)可与ERα结合,并作为ERα招募其他靶向修饰增强子与启动子相关激活酶和染色质重塑复合物的媒介。ERα调节蛋白中的共抑制因子有核受体协同抑制因子(nuclear receptor corepressor,NCoR)、配体依赖性协同抑制因子(ligand-dependent corepressor,LCoR)等,这些因子可以下调E2抑制基因的表达水平;其他共调节蛋白还包括FOXA1。研究发现,乳腺浸润性小叶癌中存在特有的FOXA1增强子结合位点,FOXA1以前馈机制调节自身表达水平,FOXA1-ER轴可以诱导TAM耐药基因的转录,靶向FOXA1增强子区域可阻断该转录程序,从而抑制浸润性小叶癌细胞的增殖水平[28]。此外,还有组蛋白和DNA修饰剂也有类似功能,如乙酰转移酶、去乙酰化酶、甲基转移酶和去甲基化酶[29-30]。ERα中存在决定转录和表观遗传活性的功能域,这些结构域是响应E2刺激的核心。雌激素信号被触发后,在LBD界面上ER单体改变构象并发生二聚化,随后ER被铰链引导至细胞核形成雌激素反应元件,最终诱导出雌激素反应[29]。表观调节因子通过调节ER启动子内CpG岛的甲基化水平,组蛋白去乙酰化酶的活性,染色质重塑、翻译后组蛋白修饰以及非编码RNA来影响肿瘤细胞的内分泌抗性。NF1缺失可能导致Ras-Raf通路的激活[31]。在一项210例乳腺癌样本测序中发现,与原发疾病相比,约8.1%的患者存在NF1靶向突变,NF1缺失促进ER非依赖性Cyclin D1的表达,体外使用CDK4/6抑制剂进行靶向治疗可抑制肿瘤进展[32]。染色质重塑也可以影响乳腺癌内分泌治疗的效果,染色质重塑复合物中的核心亚基富含AT互作结构域1A蛋白(AT rich interaction domain 1A,ARID1A)的缺失,会降低染色质的开放性,导致ER、FOXA1等先锋因子与靶染色质区域结合能力下降,ER转录活性的下降,ER阳性肿瘤细胞更易从管腔样表型转变为基底样表型,从而导致氟维司群耐药[33]。此外,非编码RNA尤其是微小RNA(microRNA,miRNA)在调控内分泌治疗耐药中也具有特殊作用,ERα阳性细胞系在使用氟维司群后miR-221/222的表达上调,导致E2耗竭减弱,肿瘤细胞获得了不依赖激素的生长途径以及对氟维司群的抗性[34]。miR-221/222还可通过激活MAPK和肿瘤抑制蛋白53(tumour protein 53,TP53)等信号通路影响肿瘤细胞对氟维司群治疗的敏感度[35]。
因此,表观遗传调节因子与内分泌治疗抵抗密切相关,其通过影响ER转录,改变ER调节因子信号网络与其他分子通路的串扰,诱导谱系分化等途径,促进了肿瘤耐药性的发生与进展。表观遗传药物作为一种强大的分子调节工具,在激活肿瘤抑制基因的同时促进ER的表达,从而阻止耐药细胞的选择和肿瘤干细胞的存活[36]。
1.4 肿瘤微环境与内分泌治疗耐药
肿瘤微环境是肿瘤产生和存活的内环境,是肿瘤与周围细胞及非细胞成分相互联系、相互影响的信号网络,与肿瘤的发生、转移,抗肿瘤的疗效与耐药性等生物学行为密切相关。近年来,越来越多的研究揭示了肿瘤免疫微环境与代谢微环境影响HR阳性乳腺癌细胞内分泌治疗效果,可导致内分泌治疗耐药。
肿瘤免疫微环境包括恶性肿瘤细胞、肿瘤相关中性粒细胞、肿瘤相关成纤维细胞、脂肪细胞和血管内皮细胞等,位于肿瘤边缘、肿瘤邻近的淋巴器官(又称三级淋巴结构)中免疫细胞的浸润水平与疾病预后密切相关[37]。研究发现,在ESR1突变肿瘤中,染色质重编程导致基底细胞角蛋白(basal cytokeratins,BCK)基因表达上调,使得ESR1突变ER阳性肿瘤细胞获得基底样特征,肿瘤可塑性增强,BCK高表达的ER阳性原发性乳腺肿瘤表现出更丰富的免疫途径,免疫激活分子信号表达增强[38]。此外,肿瘤相关巨噬细胞可通过分泌趋化因子2(chemokine,CCL2)来调节PI3K/AKT/mTOR信号轴,导致HR阳性乳腺癌细胞对他莫昔芬耐药[39]。
乳腺癌细胞通常比正常细胞摄取葡萄糖、氨基酸及脂肪酸的水平更高。大多数癌细胞依赖于有氧糖酵解和谷氨酰胺分解代谢以维持其侵略性表型。在癌细胞中,有氧糖酵解产生丙酮酸,而后大部分丙酮酸会转化为乳酸。通过多条代谢通路HR阳性乳腺癌对于他莫昔芬治疗产生内分泌抗性:如支链氨基酸可通过提高柠檬酸合成酶活性和减少线粒体活性氧,激活mTOR通路;硬脂酸、二十二碳四烯酸等可通过游离脂肪酸受体4(free fatty acid receptor 4,FFAR4)信号,从而激活AKT通路;乳酸通过乳酸脱氢酶A为癌细胞增殖提供能量,自噬活动增强[40]。此外,肥胖可促使乳腺癌细胞增殖加倍,干扰他莫昔芬和氟维司汀的抗增殖作用,脂肪代谢相关因子可能通过PI3K/AKT/mTOR和MAPK信号通路增强非基因组性ERα串扰,诱导内分泌治疗耐药[41]。
2 激素受体阳性乳腺癌靶向治疗药物
2.1 细胞周期蛋白依赖性激酶4/6抑制剂
CDK4/6抑制剂如哌柏西利、瑞波西利或阿贝西利,可以通过抑制其下游的信号传导来阻断细胞周期。在HR阳性/HER2阴性乳腺癌治疗国内外指南中,均推荐对于管腔型乳腺癌患者内分泌治疗进展后,首选CDK4/6抑制剂联合内分泌治疗。
基于PALOMA-1临床研究结果,FDA于2015年2月批准哌柏西利上市[42]。随后在CDK4/6抑制剂联合芳香化酶抑制剂Ⅲ期临床试验中,哌柏西利、瑞波西利、阿贝西利均表现出改善PFS的临床疗效,奠定了AI联合CDK4/6的一线治疗地位。一项全球多中心、随机、双盲、安慰剂对照的Ⅲ期临床PALOMA-2研究结果显示,与来曲唑单药治疗相比,哌柏西利+来曲唑组PFS[24.8个月vs14.5个月;风险比(hazard ratio,HR)= 0.56;P<0.001]和客观缓解率(ORR,55.3%vs44.4%)均有改善[43-44]。Ⅲ期临床研究MONALEESA-2对瑞波西利+来曲唑一线疗法的评估结果显示,瑞波西利+来曲唑组较安慰剂组中位PFS(25.3个月vs16.0个月;HR = 0.57;P<0.001)有所改善[45]。MONARCH 3临床研究显示,阿贝西利+AI可显著延长既往未接受系统治疗的晚期乳腺癌患者的PFS(28.2个月vs14.8个月;HR = 0.54;P<0.001)和ORR(61%vs46%)[46-47]。
CDK4/6抑制剂联合氟维司群的Ⅲ期临床试验也取得了进展。PALOMA-3研究报告了在氟维司群基础上联合哌柏西利后PFS得到显著改善(9.5个月vs4.6个月;P<0.01),总生存期(overall survival,OS)从28.0个月增至34.9个月,但无统计学意义(HR = 0.81;P= 0.09)[48-49]。MONARCH-2试验中,对比氟维司群联合或不联合阿贝西利治疗内分泌进展后的HR阳性/HER2阴性转移性乳腺癌,结果发现,氟维司群联合阿贝西利可显著改善中位PFS(16.4个月vs9.3个月;HR = 0.54;P<0.000 1)和中位OS(46.7个月vs37.3个月;HR = 0.76;P= 0.014)[50-51]。MONALEESA-3临床研究也报告了积极的结果,即加入瑞波西利联合氟维司群的一线或二线治疗可提高中位PFS(20.6个月vs12.8个月;HR = 0.59;P<0.001)[52-53]。
MONALEESA-7试验纳入均为绝经前/围绝经期乳腺癌患者,也是唯一纳入使用卵巢功能抑制剂患者的临床研究,其结果证实了瑞波西利在与非甾体类芳香化酶抑制剂/他莫昔芬+戈舍瑞林联用可使患者获益,与单独内分泌治疗相比,瑞波西利+内分泌治疗的中位PFS(23.8个月vs13.0个月;HR =0.55;P<0.001)和OS(58.7个月vs48.0个月;HR = 0.76)均得到改善[54-57](见表1)。
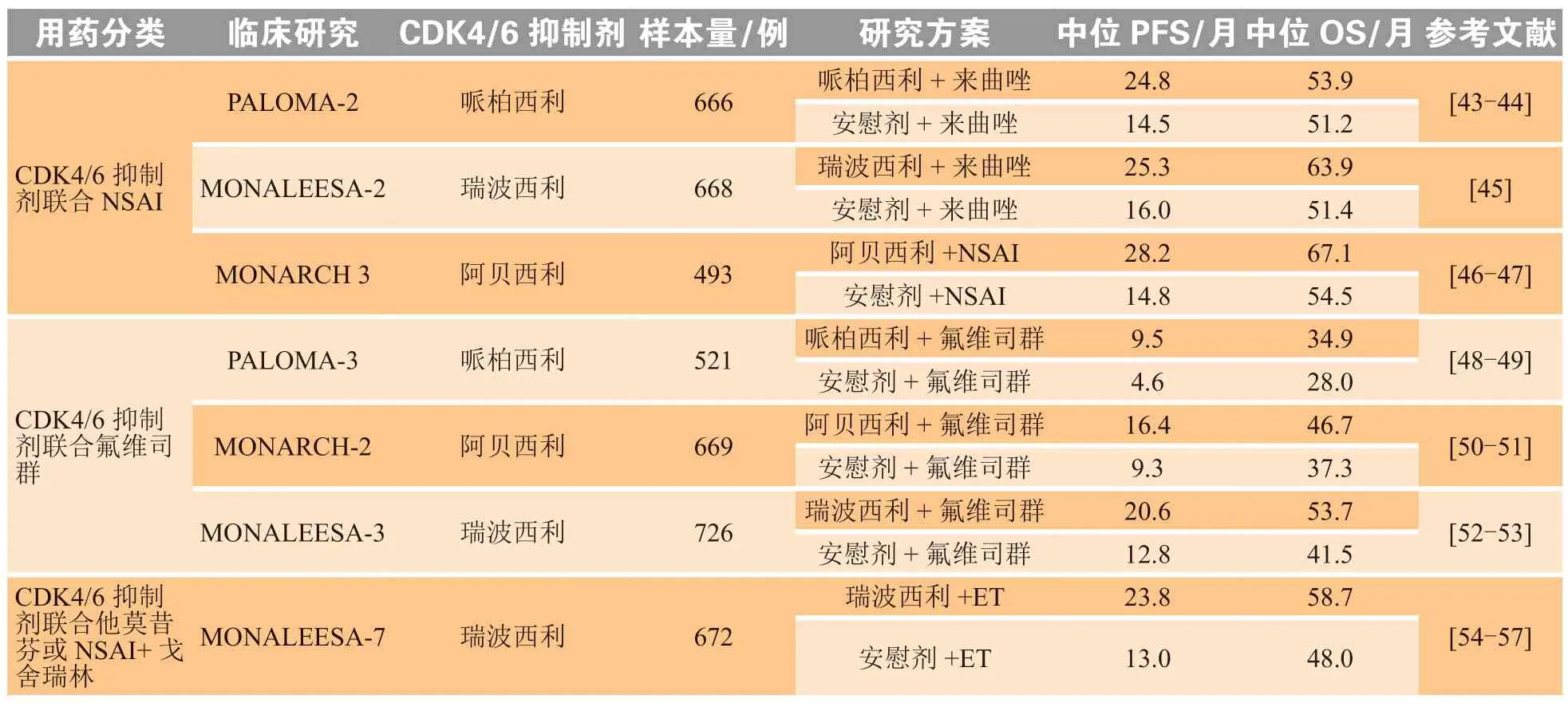
表1 CDK4/6抑制剂临床研究情况Table 1 Progress of clinical research on CDK4/6 inhibitors
2.2 PI3K/AKT/mTOR通路抑制剂
针对PAM信号通路中的突变基因,已有数种药物进入了临床研究,例如PI3K抑制剂alpelisib、inavolisib和AKT抑制剂ipatasertib、capivasertib。尽管这些药物研究对于内分泌耐药后乳腺癌展现出了一定的治疗效果,但alpelisib仅适合PIK3CA突变患者,且未在国内获批,capivasertib也还未在国内外获批适应证。
目前,mTOR抑制剂在国内有更广泛的应用基础,最典型的是依维莫司。BOLERO-2、PrE 0102等临床研究证实,以依维莫司为基础的二线联合方案具有临床疗效[58]。国际多中心Ⅲ期临床研究BOLERO-2共纳入724例患者,其研究为内分泌治疗耐药乳腺癌患者打开了新的治疗窗口,结果显示依维莫司联合依西美坦在治疗绝经后HR阳性/HER2阴性晚期、AI后复发或进展的乳腺癌患者中可延长中位PFS(7.8个月vs3.2个月;HR =0.45)[59-60]。BOLERO-5研究纳入了159例中国患者,与BOLERO-2研究结果类似,依维莫司联合依西美坦组较单用依西美坦组延长了中位PFS(7.4个月vs2.0个月;HR = 0.52),该研究为绝经后ER阳性/HER2阴性乳腺癌患者提供了新治疗选择[61]。基于CDK4/6抑制剂在二线治疗中的强势地位,TRINITI-1研究探索了依维莫司、瑞波西利、依西美坦三药联合在CDK4/6抑制剂治疗后进展的HR阳性/HER2阴性晚期乳腺癌患者中的疗效,研究结果显示,三药联合方案在24周结束时的临床获益率为41.1%,总体人群中位PFS达到5.7个月[62]。
对于绝经前HR阳性/HER2阴性晚期乳腺癌,MIRACLE研究纳入了199例国内多中心的乳腺癌患者,结果显示,依维莫司联合来曲唑组对比来曲唑组的ORR分别为50.0%和39.3%,中位PFS分别为19.4个月和12.9个月[63](见表2)。
2.3 组蛋白去乙酰化酶抑制剂
表观遗传可导致癌细胞耐药,组蛋白乙酰化和去乙酰化表达失衡可能引起乳腺癌的转移。研究表明,组蛋白去乙酰化酶(histone deacetylase,HDAC)抑制剂可诱导组蛋白赖氨酸残基乙酰化,导致肿瘤抑制基因位点的染色质重塑,从而抑制肿瘤发展[64]。
西达本胺是针对HDAC第I类、HDAC第Ⅱb类不同亚型的选择性抑制剂。在国内多中心、随机、双盲、安慰剂对照的Ⅲ期ACE临床研究中,患者随机分为西达本胺联合依西美坦组或安慰剂联合依西美坦组,研究结果表明,使用西达本胺对比安慰剂的中位PFS分别为7.4个月和3.8个月(HR =0.75),ORR分别为18.4%和9.1%,临床获益率分别为46.7%和35.5%[65]。基于此,2019年该药在国内获批用于治疗HR阳性/HER2阴性、绝经后内分泌治疗复发或进展的晚期乳腺癌。
恩替诺特是HDAC1和HDAC3选择性抑制剂,在美国开展E2112的Ⅲ期临床研究中,共招募HR阳性/HER2阴性且经AI治疗失败后转移性乳腺癌患者608例,将患者随机分为依西美坦联合恩替诺特组或安慰剂治疗组,中位PFS分别为3.3个月和3.1个月(HR = 0.87;P= 0.30),中位OS分别为23.4个月和21.7个月(HR = 0.99;P= 0.94),表明了依西美坦联合恩替诺特不能改善AI耐药后患者的预后[66]。在随后的EOC103临床研究中,恩替诺特联合依西美坦组对比安慰剂联合依西美坦组的中位PFS分别为6.3个月和3.7个月,差异有统计学意义[67]。上述2项研究的结果不一致,可能与人群基线差异、HDAC亚型表达量、恩替诺特的暴露量和疗效随访间隔时间等因素有关[66]。由于恩替诺特Ⅲ期临床研究的证据不足,目前尚未被指南纳入作为内分泌耐药后的治疗策略(见表3)。
3 激素受体阳性乳腺癌新型靶向药物
3.1 抗体药物偶联物
抗体药物偶联物(antibody drug conjugation,ADC)是近年研发出的新型抗肿瘤药物,由连接子将单克隆抗体与载药拼接起来。ADC上的单克隆抗体可以与肿瘤表面特异性靶抗原结合,以受体介导的内吞作用进入肿瘤细胞胞内,形成早期内体,快速释放载药;也可以成熟为晚期内体与溶酶体融合再释放载药,通过抑制微观聚合或DNA组装,最终导致肿瘤细胞死亡[68]。在乳腺癌领域,ADC药物取得了令人瞩目的临床研究进展,常见的研究靶点包括滋养层细胞表面抗原2(hrophoblast cell surface antigen 2,Trop-2)、HER2、HER3、脊髓灰质炎病毒受体4、受体酪氨酸激酶样孤儿素受体2等。针对晚期HR阳性/HER2阴性的乳腺癌患者,目前研究最为深入的靶点是HER2和Trop-2。
恩美曲妥珠单抗(trastuzumab emtansine,T-DM1)和德曲妥珠单抗(trastuzumab deruxtecan,T-DXd)是靶向HER2的ADC药物,其抗体部分为曲妥珠单抗,药物抗体比分别是3.5和8。在Ⅲ期临床DESTINY-Breast04研究中,共纳入了557例既往接受过内分泌和一线/二线化疗的HR阳性或HR阴性、HER2低表达的转移性乳腺癌患者,在HR阳性患者中T-DXd组对比医生选择治疗组(treatment of physician,s choice,TPC),TPC组中的化疗方案包括艾力布林、卡培他滨、白蛋白-紫杉醇、吉西他滨和紫杉醇,结果显示,T-DXd组和TPC组的中位OS分别为23.9个月和17.5个月(HR = 0.64),中位PFS分别为10.1个月和5.4个月(HR = 0.51),T-DXd组的疗效较好且总体安全性可控[69]。
戈沙妥珠单抗(SG)、Dato-DXd和SKB264均是靶向Trop-2的ADC药物,Ⅲ期TROPiCS-02研究纳入了543例HR阳性/HER2阴性转移性乳腺癌患者,SG与TPC相比显著改善了中位PFS(5.5vs4.0个月;HR = 0.66;P= 0.000 3),在中位OS上也显示出优势(14.4 个月vs11.2个月;HR = 0.79;P=0.02)[70-71]。Ⅲ期TROPION-Breast01研究结果显示,Dato-DXd组与化疗组相比提升了既往经治HR阳性/HER2阴性转移性乳腺癌患者的PFS(6.9个月vs4.9个月;P<0.000 1)[72]。SKB264的Ⅱ期研究结果也表现出较好的抗肿瘤效果,中位随访时间为8.2个月,ORR为36.8%,中位PFS为11.1个月[73]。
综上所述,以T-DXd为代表的系列ADC类药物为CDK4/6抑制剂经治进展后的晚期乳腺癌患者带来更多治疗选择,未来ADC药物的研发在提高靶向性,增强连接子稳定性,改善耐药等方面还有巨大的探索空间。
3.2 细胞周期抑制剂
近年来,研究者们对细胞周期蛋白进行了深入探索。除了CDK4和CDK6,CDK7也是细胞周期中的一个关键激酶,既可阻断CDK1 ~ CDK4、CDK6在细胞周期中的调控作用,又可影响致癌基因的转录过程。在CDK7抑制剂CT7001联合氟维司群治疗HR阳性/HER2阴性晚期乳腺癌的临床试验中,入组的31例患者均经CDK4/6抑制剂治疗且疾病出现进展,在疗效可评估的24例患者中,71%的患者肿瘤缩小,8%达到部分缓解,另有54%的患者显示疾病稳定[74]。另一种高效、口服CDK7抑制剂SY-5609则在复发转移性胰腺癌患者中疗效显著[75],被FDA授予孤儿药资格。多项针对晚期恶性实体瘤的国产口服CDK7抑制剂的Ⅰ/Ⅱ期临床研究正在多中心开展。第1、2代CDK抑制剂存在靶向性有限、毒副作用严重的问题,抗肿瘤疗效有限,随着以哌柏西利为代表的第3代CDK抑制剂在临床上崭露头角,CDK8/19、CDK9、CDK12等多种CDK抑制剂均处于Ⅰ/Ⅱ期临床研究中。由于泛CDK抑制剂的治疗窗不足,未来通过提高选择性CDK抑制剂的安全性,改善耐药,其在晚期乳腺癌后线治疗中还可发挥更大的临床价值。
4 结语与展望
乳腺癌内分泌治疗耐药的形成机制复杂,可能与ESR1突变、CYP19A1扩增,PAM、RTK、MAPK肿瘤相关信号通路有关,也可能受到肿瘤微环境,如肿瘤相关巨噬细胞、脂代谢等的影响。新型耐药靶点与药物的开发持续改善了HR阳性乳腺癌的预后。延长药物治疗周期,改善耐药性可有效提高患者的生存质量,改善生存结局。近年来,晚期HR阳性乳腺癌的治疗已形成了规范的体系。CDK4/6抑制剂与内分泌治疗药物联用是HR阳性内分泌治疗耐药后的首选方案,针对HER2和Trop-2靶点的新型ADC药物为HR阳性/HER2阴性及HER2低表达患者带来了福音。未来可通过挖掘更多治疗靶点,提高HR阳性乳腺癌亚组人群治疗的敏感性,从而扩宽内分泌治疗和靶向治疗的覆盖领域。
