基于磁性分子印迹技术检测食品中利巴韦林含量
2024-03-19金文丽周鑫魁李建民张连钢张婧蕾袁嘉男
金文丽,周鑫魁,李建民,张连钢,张婧蕾,袁嘉男
1.嘉峪关市食品药品和医疗器械检验检测中心 (嘉峪关 735100);2.甘肃省商业科技研究所有限公司 (兰州730010)
利巴韦林为白色结晶性粉末,无臭。易溶于水[1-2]。被用于治疗病毒性引起的肝炎,腮腺炎,肺炎[3]。利巴韦林服用之后会引起神经系统反应,比如眩晕,还会导致消化系统反应,出现恶心、消化不良等症状[4]。违法商家会将其添加在殖饲料中,减少养殖过程病毒的侵害[5]。消费者会购买并食用带有抗生素的肉制品,从而对人体健康产生潜在性的危害。国家标准规定肉及肉制品中不得检出利巴韦林[6]。利巴韦林的检测,需要经过酸化提取,硼酸固相萃取柱净化,液相色谱质谱联用仪检测。在实际检测试验中,实验操作要求极高,微小的偏差都会影响利巴韦林的检测结果[7-8]。所以建立一种准确性高、稳定性好、易操作的检测方法是有必要的。
分子印迹是通过印迹分子与模板分子之间通过π键和非共价键结合。生成的印迹聚合物[9-10]。分子印迹微球技术,将聚合物微球经过研磨,得到粒径均一的分子印迹吸附材料[11]。可用于固相萃取过程中。本研究旨在利用自主合成磁性分子印迹材料[12],建立一种稳定性良好的快速,高效,准确,易操作的利巴韦林检测方法。现有报到中,很少由关于磁性分子印迹材料用于食品中利巴韦林的检测的实验。研究也为利巴韦林检测方法提供新的思路,为磁性分子印迹拓宽应用领域打好基础。
1 材料与方法
1.1 仪器和试剂
7307振动样品磁强计(美国Lake Shore公司);H-800透射电镜(日本日立公司);岛津20A液相色谱仪(日本岛津科技有限公司);IRSpirit红外光谱仪(日本岛津科技有限公司)。
利巴韦林(化学纯,南京都莱生物技术有限公司);氯化铁,分析纯、氯化亚铁(分析纯,天津市百世化工有限公司);α-甲基丙烯酸(MAA,分析纯,阿拉丁试剂上海有限公司);三羟甲基丙烷三甲基丙烯酸酯(TRIM,分析纯,阿拉丁试剂上海有限公司);偶氮二);硅烷偶联剂KH570(MPS,分析纯,阿拉丁试剂上海有限公司);甲苯(分析纯,天津市化学试剂公司);正硅酸乙酯(TEOS,分析纯、天津光复科技发展有限公司);聚乙烯醇2000(分析纯、天津光复科技发展有限公司);盐酸(分析纯、天津光复科技发展有限公司);甲醇(色谱纯,北京百灵威科技有限公司);乙腈(色谱纯,北京百灵威科技有限公司);去离子水(用优普超纯水制备仪制备);肉及肉制品样品(购于超市及农贸市场)。
1.2 方法
1.2.1 Fe3O4磁性化合物制备
参考文献[13]方法,利用共沉淀法制备磁性Fe3O4。称取4.7 g氯化铁和1.9 g氯化亚铁于300 mL球形,可加热,耐压反应釜中。加入120 mL去离子水,通入氮气20 min,加入10.5 mL氨水,继续通入20 min氮气,密封反应釜。将反应釜加热至内部温度105 ℃,反应1 h。反应结束,冷却至常温,用强磁计在反应釜外部吸附生成的Fe3O4磁性化合物,将上层反应残留液倾倒出反应釜,并反复加入去离子水洗涤Fe3O4磁性化合物,直至洗涤液变为中性。收集Fe3O4磁性化合物与干燥皿中,于60 ℃真空干燥6 h。得到黑色粉末即为Fe3O4磁性化合物。
1.2.2 含Fe3O4磁性化合物聚合单体制备
参考文献[14]方法,以TEOS为硅源,碱催化溶胶-凝胶法,制备Fe3O4磁性化合物聚合单体。称取1.0 g 1.2.1小节中制备的Fe3O4磁性化合物于500 mL磨口三角瓶中,加入200 mL乙醇,超声使Fe3O4磁性化合物分散于乙醇中。快速搅拌并依次快速加入3 mL去离子水、8 mL氨水和9 mL TEOS,迅速盖紧瓶盖。于电磁搅拌加热器上,转速300 r/min于,70 ℃反应5 h。用强磁计在三角瓶外部吸附生成产物,倾倒出全部上层残留液体,并反复加入去离子水洗涤产物,直至洗涤液变为中性。向瓶中加入200 mL乙醇,超声使其分散。快速搅拌并加入12 mL MPS,在温度40 ℃、转速300 r/min条件下搅拌反应8 h,反应产物经外磁场分离,并用去离子水多次洗涤。最后,将产物于60 ℃真空干燥6 h。得到黑色粉末,即为Fe3O4磁性化合物聚合单体。
1.2.3 利巴韦林磁性分子印迹微球(MMIPs)制备
参考文献[15]方法。在三口反应瓶中加入0.32 g利巴韦林,0.1 g AIBN,0.02 g聚乙烯醇和120 mL去离子水。充入20 min氮气,搅拌下升温至65 ℃使其完全溶解。加入0.2 g 1.2.2小节中制备的Fe3O4磁性化合物聚合单体,持续通入氮气,以1滴/3 s的滴速由加液漏斗滴加0.45 mL MAA和14.0 mL TRIM,10.0 mL苯的混合溶液,滴加结束后,持续通氮气,搅拌下升温至70 ℃反应7 h。聚合反应结束后将得到的聚合物微球滤出,依次用蒸馏水、甲醇、丙酮各100 mL洗涤。除去洗掉苯、剩余的有机单体等,按甲醇-乙酸体积比70∶30索氏抽提48 h,洗去分子模板,用去离子水将分子印迹清洗干净。直至清洗液液用紫外检测不到模板分子,在50 ℃下真空干燥24 h,放入干燥器备用。磁性非分子印迹微球(MNIPs)的制备与MMIPs相同,但不加入利巴韦林。
1.2.4 利巴韦林液相色谱检测条件
色谱条件:a)色谱柱4.6×150 mm,C18键合填料色谱柱;b)流动相为50 mmol/mL的pH 4.0乙酸铵缓冲液+乙腈;c)流速为1 mL/min;d)柱温为30 ℃;e)进样量为20 μL;f)波长为230 nm。
1.2.5 利巴韦林MMIPs和MNIPs吸附容量测定
分别称取7份0.01 g MMIPs和MNIPs置于10 mL离心管中,7支离心管中分别加入5.00 mL不同质量浓度的利巴韦林乙腈溶液,其质量浓度分别为1,2,5,10,20,30和50 μg/mL。在常温下涡旋振荡1 min,用强磁计操作进行利巴韦林MMIPs和MNIPs与溶液分离,并通过液相色谱检测残留乙腈溶液中利巴韦林的含量。按式(1)计算MMIPs和MNIPs对利巴韦林的吸附容量。并绘制静态吸附等温线。
式中:Q为吸附容量,mg/kg;C初始为吸附前利巴韦林初始质量浓度,μg/mL;C残留为吸附后利巴韦林残留质量浓度,μg/mL;V为溶液体积,mL;m为MMIPs和MNIPs的使用质量,g。
分别称取7份0.01 g MMIPs和MNIPs置于10 mL离心管中,7支离心管中分别加入5.00 mL质量浓度为30 μg/mL的利巴韦林乙腈溶液。在常温下,分别涡旋振荡10 s,20 s,30 s,45 s,1 min,2 min和5 min,用强磁计操作进行利巴韦林MMIPs和MNIPs与溶液分离,并通过液相色谱检测,残留乙腈溶液中利巴韦林的含量。按式(1)计算MMIPs和MNIPs对利巴韦林的吸附容量。并绘制吸附动力学曲线。
1.2.6 利巴韦林MMIPs专一性验证
利巴韦林结构为1-β-D-呋喃核糖基-1H-1,2,4-三氮唑-3-羧酰胺,现将利巴韦林,阿昔洛韦,青霉素,四环素等抗菌或抗病毒药物混合,配制成溶液。其质量浓度分别为利巴韦林10 μg/mL,阿昔洛韦10 μg/mL,青霉素10 μg/mL,四环素10 μg/mL。分别称取4份0.01 g MMIPs置于盛有上述溶液的10 mL离心管中,在常温下,涡旋振荡1 min,用强磁计操作进行利巴韦林MMIPs与溶液分离,并通过液相色谱检测残留溶液中利巴韦林的含量。并参考文献[18]方法检测残留阿昔洛韦的含量,参考文献[19]方法检测残留青霉素的含量,参考文献[20]方法检测残留四环素的含量。
1.2.7 MMIPs对利巴韦林的吸附pH条件
在不同pH环境下,分子印迹对目标物质利巴韦林的吸附性质也有所不同。分别用pH为2.0,3.0,4.0,5.0,6.0,7.0,8.0,9.0,10.0和11.0的乙腈缓冲液,配制10 μg/mL的利巴韦林溶液。分别取上述溶液5.00 mL于10 mL离心管中。按照1.2.5小节处理,并进入高效液相色谱仪测定利巴韦林的含量。
1.2.8 利巴韦林溶液中溶剂对吸附效果的影响
目标物被吸附在MMIPs上,可以用甲醇、甲醇-水溶液、纯水将其从分子印迹上洗脱下来,所以上样溶液不能是含纯甲醇或者含水的溶液。一定要选择合理的溶液比例,确保MMIPs对目标物达到最佳吸附条件。选择体积乙腈体积分数为0,5%,10%,15%,20%,30%,40%,50%,60%,70%,80%,90%和100%的乙腈-水溶液,配制10 μg/mL的利巴韦林标准溶液。分别取上述溶液5.00 mL于10 mL离心管中。按1.2.5小节处理,并进入高效液相色谱仪测定残留液中利巴韦林的含量,用强磁计操作进行利巴韦林MMIPs与溶液分离,用5 mL去离子水洗涤MMIPs,收集全部洗涤液,进入液相色谱检测。
1.2.9 固相分散萃取填料的制备
称取30 g MMIPs,45 g PSA,45 g C18和60 g无水硫酸钠,混合后,于均质机中均质混匀。得到灰黑色固相分散萃取填料。
1.2.10 样品前处理
肉及肉制品一般为固体。其中含有大量蛋白质、油脂等杂质,会对液相色谱、液相质谱检测结果造成影响。需要建立一种方法,既能有效地从样品中提取目标物利巴韦林,又能减少杂质的影响。由于利巴韦林易溶于水、较易溶于甲醇、乙腈,所以可以用乙腈直接提取经分子印迹固相萃取填料净化富集,用水洗脱,进行检测。
称取5.00 g粉碎后的样品于50 mL离心管中,加入20 mL乙腈提取,乙腈可以使样品蛋白质变性沉淀。涡旋2 min后,加入2 g无水硫酸钠,涡旋振荡1 min后,在2 000 r/min条件下离心2 min。取全部上清液于新的50 mL离心管中,加入1.0 g固相分散萃取填料,涡旋振荡1 min,用强磁计操作进行利巴韦林MMIPs于溶液分离。用1 mL乙腈洗涤离心管底部黑色固体粉末,弃去乙腈,用1 mL去离子水洗涤离心管底部黑色固体粉末,收集全部洗涤液,经过0.22 μm滤膜后,进入液相色谱检测。并验证实际样品检测中的回收率,精密度,检出限,线性范围。
2 结果与讨论
2.1 表征
2.1.1 MMIPs电镜表征
由图1可知,所制备的MMIPs呈球形,微球平均粒径为2.5 μm。从电镜图中可以看出,微球粒径均匀,且没有其他不规则形状。说明制备的材料单分散性良好。材料具有单分散性,有利于后续实验中目标物的吸附和解析。同样增加材料的使用稳定性。材料中加入Fe3O4磁性化合物聚合单体,该单体中有SiO2,增加材料刚性。后续实验中该材料在离心,涡旋,超声条件下仍保持良好的机械强度,确保吸附性能的稳定。

图1 MMIPs透射电镜图
2.1.2 MMIPs红外表征
由图2可知,Fe3O4磁性化合物聚合单体,915 cm-1处为聚合物单体的环氧基伸缩振动吸收峰,1 097 cm-1处出现的谱带为SiO2的不对称伸缩振动吸收,1 635 cm-1为C=C的伸缩振动峰。经聚合反应,得到MMIPs微球,C=C参与反应,该处吸收峰消失。2 920 cm-1和2 850 cm-1处出现了—CH2—的不对称伸缩振动峰和对称伸缩振动峰,说明发生了聚合反应。

图2 MMIPs红外光谱图
2.2 利巴韦林液相色谱图
由图3可知,利巴韦林在2.248 min出现特征色谱峰,峰型良好,没有严重的前延和拖尾现象,周围没有杂质峰影响定量积分。证明建立的样品前处理条件能达到净化目的,色谱条件能达到分离定量及定性的目的。
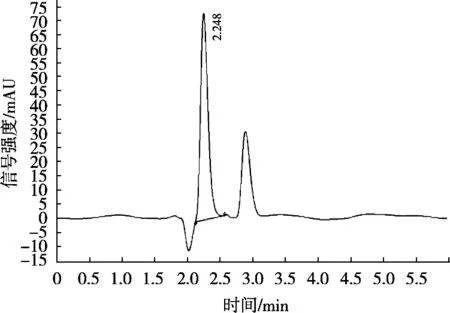
图3 利巴韦林标准及样品色谱图
2.3 利巴韦林MMIPs和MNIPs吸附容量
以MMIPs和MNIPs对利巴韦林的吸附容量为纵坐标,吸附母液浓度为横坐标,绘制的静态吸附等温线如图4(a)所示。以MMIPs和MNIPs对利巴韦林的吸附容量为纵坐标,吸附时间为横坐标,并绘制吸附动力学曲线如图4(b)所示。
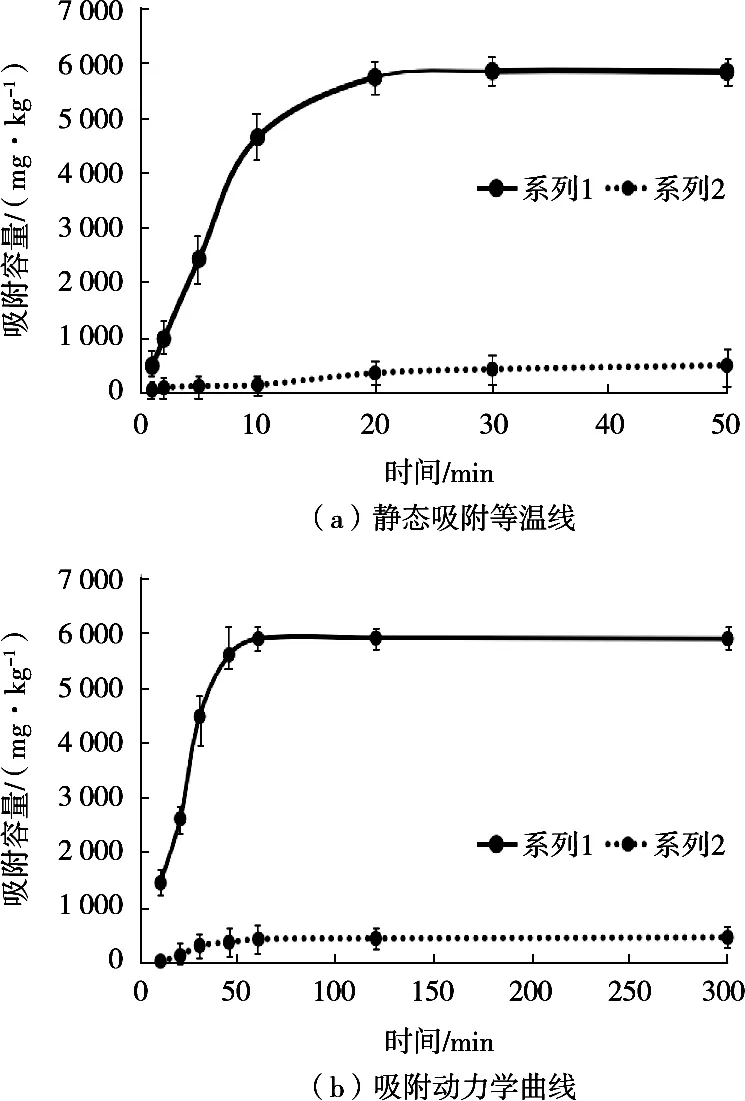
图4 MMIPs和MNIPs对利巴韦林的吸附等温线(a)和吸附动力学曲线(b)
由图4(a)可知,MMIPs的吸附容量随利巴韦林浓度增加而增大,但是当目标物质量浓度超过10 μg/mL时,吸附容量增大趋势缓慢,当吸附容量超过5 835 μg/g时,达到最大吸附容量,随利巴韦林浓度增加,吸附容量几乎不变。是因为MMIPs在制备过程中形成的有效吸附点位的数量有限,无法无限制的吸附目标物。由图4(b)可知,当吸附时间的增加,吸附容量也随之增大,在涡旋振荡1 min内达到吸附平衡,继续增加吸附时间,吸附容量几乎不增大。此处可以看出本研究制备的MMIPs能在极短的时间内完成对目标物的吸附。较文献[20]中报到的40 min达到吸附平衡,极大的提高了吸附效率。这是因为MMIPs微球,在制备过程中形成的2 μm多孔单分散微球,微球比表面积大,与目标物作用速度快。这也为后续检测方法的建立奠定基础。而MNIPs对利巴韦林的几乎不产生吸附效果。也说明MMIPs的制备是成功的。
2.4 利巴韦林MMIPs专一性
MMIPs对利巴韦林,阿昔洛韦,青霉素,四环素的吸附容量如表1所示。由表1可知,MMIPs对利巴韦林吸附效率大于95%,10 μg/mL质量浓度下吸附容量为4 975 mg/kg。而MMIPs阿昔洛韦、青霉素、四环素的吸附效率不超过2%,吸附容量不超过65 μg/g,可以认为MMIPs专一性良好,对利巴韦林以外的药物几乎不产生吸附效果,这是因为试验中四种药物虽然都是抗菌抗病毒的药物,结构也有一定程度上的相似性,但是分子大小和空间结构,仍然存在很大差异,与MMIPs吸附位点不完全匹配,所以其他分子的吸附容量很低。
2.5 MMIPs对利巴韦林的吸附pH条件
以pH为横坐标,残留液中利巴韦林浓度为纵坐标,得到不同pH对MMIPs吸附利巴韦林的影响趋势图,如图5所示。
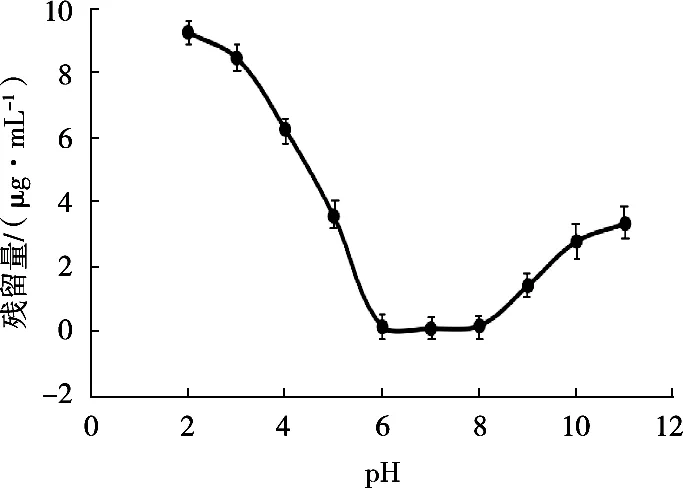
图5 溶液pH对吸附效果的影响
由图5可知,当pH<6或者pH>8时,分子印迹对利巴韦林的吸附有明显下降趋势。而在6≤pH≤8范围内,分子印迹对利巴韦林的吸附并不受pH影响。所以前处理使用乙腈提取,保持提取液,上样液均为中性,满足pH要求。
2.6 利巴韦林溶液中溶剂对吸附效果的影响
以溶液中乙腈含量为横坐标,残留液和洗脱液中利巴韦林的含量为纵坐标绘制曲线图,得到溶剂对MMIPs吸附和洗脱效果的影响趋势,如图6所示。
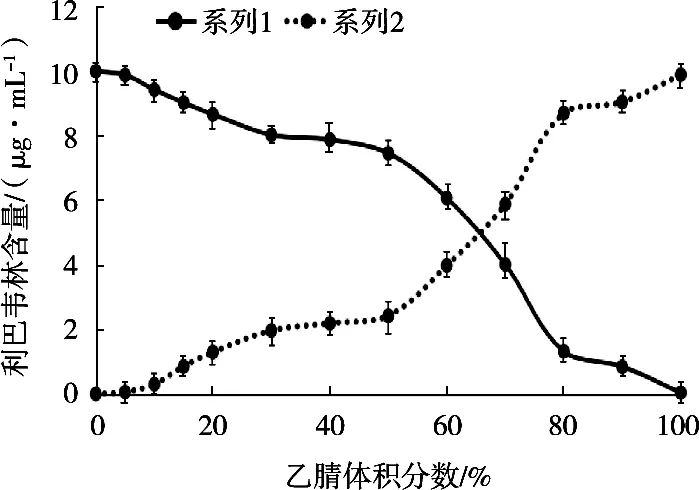
图6 溶液中乙腈含量对吸附和解析效果的影响
由图6可知,当水含量越大时,测得利巴韦林最终含量逐渐减小,而残留液中目标物含量逐渐增加。说明,当溶液中水含量增加时,MMIPs对目标物的吸附能力减弱,目标物仍然留在溶液中,影响最终检测结果。当溶液为纯乙腈时,MMIPs可以有效的吸附5 mL溶液中的目标物。而考虑乙腈提取样品时可以有效沉淀样品中大部分蛋白质。所以选择乙腈为提取液进行试验。这是因为,利巴韦林溶解性导致,利巴韦林结构中含有一个核糖分子,极易溶于水。水对利巴韦林的溶解性,强于MMIPs对利巴韦林的吸附性。而乙腈对目标物的溶解性弱于MMIPs的吸附性,所以可以利用乙腈提取目标物,用水洗脱目标物。这样的现象增加了实际样品检测方法建立的可行性。
2.7 方法加标回收率及方法稳定性实验
在阴性样品中分别加入标准溶液,使得样品中目标物含量为0.5,2.0和10.0 mg/kg,每个浓度水平,平行三组试验,按照1.2.10小节方法处理样品,并进行检测,结果见表2。

表2 三水平加标回收率及精密度实验结果
由表2可知,此方法对样品中利巴韦林检测的回收率在80.6%~90.2%之间,SRSD为4.38%,满足检测要求。该方法能达到回收率高、精密度好的效果,是因为分子印迹材料对目标物吸附能力强,富集效果好,选择良好的洗脱试剂,可以快速高效地洗脱分子印迹固相萃取中的目标物。
2.8 重复性
取阳性样品,按照1.3.10小节方法处理,进行检测。重复8次试验,以每次色谱峰峰面积的相对标准偏差计算定量重复性,以每次色谱峰出峰时间的相对标准偏差计算定性重复性,结果见表3。
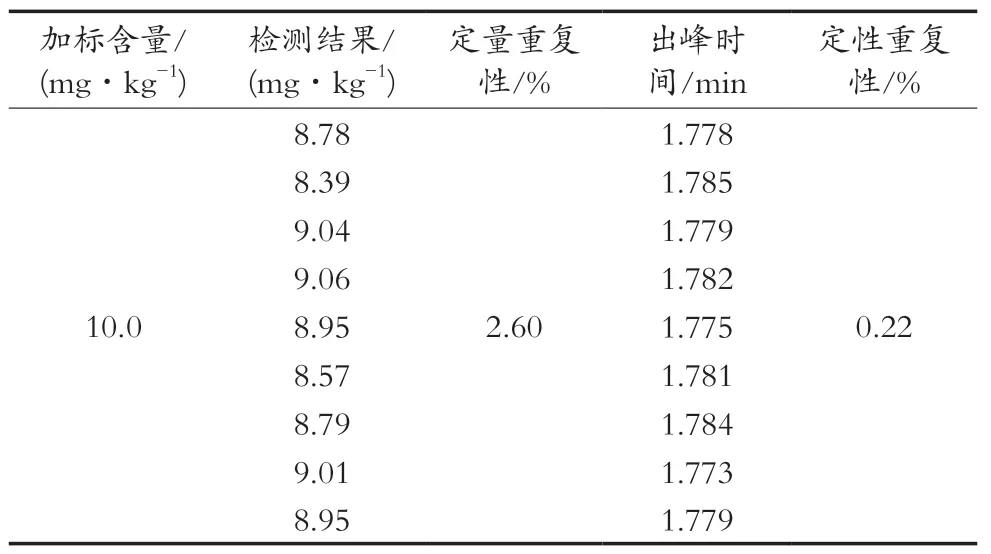
表3 重复性实验结果
SRSD(定性)=0.22%,SRSD(定量)=2.60%。均小于3%,检测结果稳定,满足检测要求。由于分子印迹对利巴韦林吸附速度快,短时间内可以达到吸附平衡,且吸附效率很高,影响吸附效果的因素较少。在相对稳定的实验条件下,所以得到的检测实验结果相对稳定。而且分子印迹没有吸附样品中可能影响利巴韦林色谱行为的杂质,所以在色谱分离和检测过程中,利巴韦林可以在特定的时间范围内出现色谱峰。
2.9 检出限及线性范围
以30 min内基线的3倍噪声对应的浓度为仪器最低检出限,以仪器检出限按照方法稀释倍数计算方法检出限,以3倍检出限浓度对应的浓度为定量限。以30 min内基线的3倍噪声对应的浓度为仪器最低检出限,检出限为0.05 μg/mL。将仪器检出限带入方法中计算该方法的方法检出限,方法检出限为0.02 μg/g,定量限为0.05 μg/g,适用于微量检测。分别配制0.1,0.5,1.0,5.0,20.0和50.0 μg/mL利巴韦林标准溶液,以1.3.4小节的色谱条件进行检测。得到不同浓度标准溶液对应的色谱峰面积,以浓度为横坐标,面积为纵坐标,检测结果在0.1~50.0 μg/mL范围内呈良好线性,线性回归方程y=541.33x+12.092;相关性系数R2=0.999 8,满足检测要求。
3 结论
在成功制备了单分散性良好的磁性分子印迹微球的基础上,研究了该材料对利巴韦林的吸附容量,吸附pH条件,吸附溶剂等影响因素。以MMIPs为原料,C18、PSA、硫酸钠为辅料制备针对肉及肉制品中利巴韦林检测试验,样品处理过程使用的固相分散萃取填料。并建立实际样品中利巴韦林的检测方法。验证该方法的回收率,精密度,定量重复性,定性重复性,检出限和线性范围。均符合方法学要求,可以用于实际实验过程中。该方法操作简便,回收率高,结果稳定,样品处理耗时短,消耗有机试剂少,适合批量样品处理。
