负载3-甲基腺嘌呤介孔二氧化硅的制备与释药性能研究
2024-03-14韩卓越王秀巫业振胡浩然邢亚群蚌埠医学院第二附属医院药剂科安徽蚌埠33000蚌埠医学院药学院安徽蚌埠33000
韩卓越,王秀,巫业振,胡浩然,邢亚群(1.蚌埠医学院第二附属医院药剂科,安徽 蚌埠 33000;.蚌埠医学院药学院,安徽 蚌埠 33000)
自噬是广泛存在于真核细胞内的一种溶酶体依赖性的细胞降解途径,是机体内存在的一种自我修复和维持生命的过程。自噬可以表现出多种功能形式,包括发挥促生存作用的细胞保护形式、促进肿瘤细胞死亡的细胞毒性形式和不具有保护作用的非保护形式[1]。3-甲基腺嘌呤(3-MA)是一种非特异性磷脂酰肌醇3-激酶(PI3K)抑制剂,通常在体外用于抑制自噬[2],可与抗肿瘤药物联合发挥协同作用。研究表明早期自噬抑制剂 3-MA的使用可保护心肌细胞免受阿霉素诱导的心脏毒性[3]。3-MA 可增强顺铂耐药下咽鳞状癌细胞的化疗敏感性[4],还可促进埃索美拉唑在胃癌细胞中的抗增殖活性[5]。广泛使用3-MA可通过自噬激活显著降低氯仿对小鼠的肝毒性[6]。此外,3-MA抑制自噬能够在体外增强缺氧诱导的人结直肠癌细胞凋亡[7]。有研究已证明3-MA在体外和体内均可抑制骨肉瘤的生长[8]。可见3-MA在自噬相关的疾病中发挥了重要作用,尽管用途广泛,但 3-MA只有在高浓度时才有效,并且在室温下的溶解度很差[9]。因此,更有效、更特异性的递送自噬抑制剂对于开发基于抑制自噬协同抗肿瘤的辅助治疗方法至关重要。
在纳米技术领域中已经报道了许多纳米载体,其中,介孔二氧化硅(MSN)纳米粒子是现阶段非常理想的新型药物载体,其具有独特的多孔结构,良好的化学稳定性、表面功能性和生物相容性,能确保多种药物分子的可控释放和药物靶向性传递[10-11]。在纳米载体基础上通过靶向修饰可以改善药物利用率,提高治疗效果,并减少全身毒性相关的不良反应。骨靶向药物唑来膦酸(ZOL)是双膦酸盐的成员之一,对骨组织表现出良好的亲和力,具有与焦磷酸盐类似的调节骨骼矿化的能力,且比焦磷酸盐更稳定,其主链存在两个膦酸基,可以与羟基磷灰石表面双齿结构中的二价钙离子进行螯合[12-13]。因此,ZOL是治疗骨相关疾病靶向配体的最佳选择。本研究设计了一种将ZOL连接在聚多巴胺(PDA)包覆的MSN纳米粒上,并装载3-MA,通过性质表征及药物在不同条件下的释放行为研究,初步评价其基本性能。
1 仪器与试药
1.1 仪器
仪器 ZNCL-GS智能磁力搅拌器(上海予申仪器有限公司);5424R离心机(艾本德生命科学公司);KQ-500B型超声波清洗器(昆山市超声仪器有限公司);UV-3600紫外分光光谱仪(岛津公司);JEM-2100F透射电子显微镜(日本电子公司);IS50红外分光光谱仪(赛默飞世尔科技公司)。
1.2 试药
3-MA(批号:C13979809)、ZOL(批号:C134-76263)、十六烷基三甲基氯化铵(CTAC,批号:C12197343)、正硅酸乙酯(TEOS,批号:C12561839)、三乙醇胺(TEA,批号:20210507)、N,N-二甲基甲酰胺(DMF,批号:P2136663)、无水乙醇(批号:2211073501)、盐酸多巴胺(批号:C12659279)、二甲基亚砜(DMSO,批号:P2087852)(上海麦克林)。N,N'-羰基二咪唑(CDI,批号:K00220563)、NH2-PEG-SH(批号:K00220819)(西安凯新生物)。
2 方法与结果
2.1 纳米制剂的制备
2.1.1 MSN的制备 按照文献[14]的方法,并进行相应优化,简要方法如下:取适量CTAC置量瓶中加入去离子水(20 mL)混合均匀,加入TEA(0.06 g),在95℃油浴,搅拌1 h后逐滴加入TEOS(1.5 mL),滴加完毕后继续搅拌1 h,冷却至室温,离心收集,沉淀醇洗3次。然后在酸性乙醇溶液中60℃冷凝回流4 h,去除致孔模板剂CTAC,离心收集,乙醇洗涤两次。同样的提取工艺重复两次。离心后的沉淀冷冻干燥24 h,最终得到白色粉末MSN。
2.1.2 PDA的包覆 参考文献[15]已有的方法:将100 mg MSN完全溶解在50 mL Tris-HCl缓冲液中(pH 8.5),加入50 mg盐酸多巴胺,室温有氧条件下搅拌(200 r·min-1)4~6 h后,离心 10 min,沉淀用去离子水洗涤两次,最终产物冷冻干燥,记为MSN@PDA。
2.1.3 MSN@PDA-PEG-ZOL的制备
① 活化ZOL:将ZOL(100 mg)与适量TEA溶解在DMF中,加入CDI(90 mg,无水),密闭氮气保护,60℃油浴,搅拌反应24 h。用旋转蒸发仪蒸发未反应完的TEA。沉淀物用乙腈洗涤两次以去除多余的CDI,旋转蒸发干燥,得到纯活性沉淀物 ZOL[16]。
② 活化ZOL与NH2-PEG-SH连接:将纯活性沉淀物ZOL(22.6 mg)溶解在适量DMSO和TEA中,将分子量为2000的NH2-PEG-SH(质量浓度为2 mg·mL-1)滴加到溶液中,在密闭的氮气下反应12 h。反应混合物在蒸馏水中透析48 h以去除多余的活性ZOL,得到ZOL-PEG-SH[17]。
③ MSN@PDA-PEG-ZOL的制备:将上述所得化合物溶解在20 mL Tris-HCl缓冲液(pH 8.5)中,加入100 mg MSN@PDA,室温搅拌过夜。然后,将溶液在4℃下以12 000 r·min-1离心10 min,得到MSN@PDA-PEG-ZOL,用去离子水洗涤三次后,冷冻干燥。
2.2 3-MA的包封率(EE)及载药量(DL)的测定
2.2.1 3-MA标准曲线的建立 称取5 mg 3-MA于25 mL量瓶中,加去离子水配制200 µg·mL-1的标准溶液,取标准溶液稀释成1.56、3.125、6.25、12.5、25、50 µg·mL-1的系列工作液,使用紫外检测,波长为273 nm。以3-MA浓度(C)设为横坐标,吸光度(A)为纵坐标,建立标准曲线。结果表明3-MA在1~50 µg·mL-1与峰面积线性关系良好,回归方程为A=0.0841C+0.0159(R2=0.9995)。
2.2.2EE和DL的测定 取3-MA对照品溶解在去离子水中,分别配制50、100、200、500、1000µg·mL-1的标准溶液。取20 mg MSN 分别放在20 mL的离心管中,依次加入10 mL不同浓度的3-MA标准溶液,水浴超声至MSN完全分散,室温下搅拌24 h后,离心收集上清液,用去离子水洗涤沉淀两次。洗涤液与上清液的合并液用于载药量和包封率的测定[18];沉淀冷冻干燥后备用,记为MSN@3-MA。
取合并液适量,0.45 μm微孔滤膜过滤,用去离子水稀释至适宜浓度,测定其吸光度,代入标准曲线方程计算3-MA浓度,EE和DL分别按公式(1)、(2)计算。
式中3-MA的含量记为W1(g),MSN@3-MA称重记为W2(g),初始3-MA投入量记为W3(g)。
结果如图1和表1所示,3-MA在273 nm处存在特征峰,而MSN@3-MA的UV曲线中存在3-MA的特征峰,证明3-MA加载在纳米粒子中。不同3-MA浓度的DL和EE如表1所示,在50~1000µg·mL-1范围内,DL随浓度的增加而增加,浓度在1000 µg·mL-1时,EE为(90.87±0.05)%,DL为(37.55±0.02)%。

表1 不同浓度3-MA的EE及DLTab 1 Encapsulation rate and drug loading amount of different mass ratios of 3-MA

图1 3-MA、MSN、MSN@3-MA的UV图谱Fig 1 UV spectrum of 3-MA,MSN,and MSN@3-MA
2.3 MSN@PDA-PEG-ZOL的表征分析
2.3.1 核磁共振氢谱(1H-NMR)分析 取制得的产物适量,在DMSO中检测,检测条件为 500 MHz,利用核磁共振氢谱仪对样品进行测定。结果如图2 所示,ZOL结构中相应氢的位置见图2A,羟基活泼氢在谱图中未出峰,图2B中ZOLPEG-SH氢谱图出现与ZOL结构中相应氢的位置一致,证实两药合成成功。
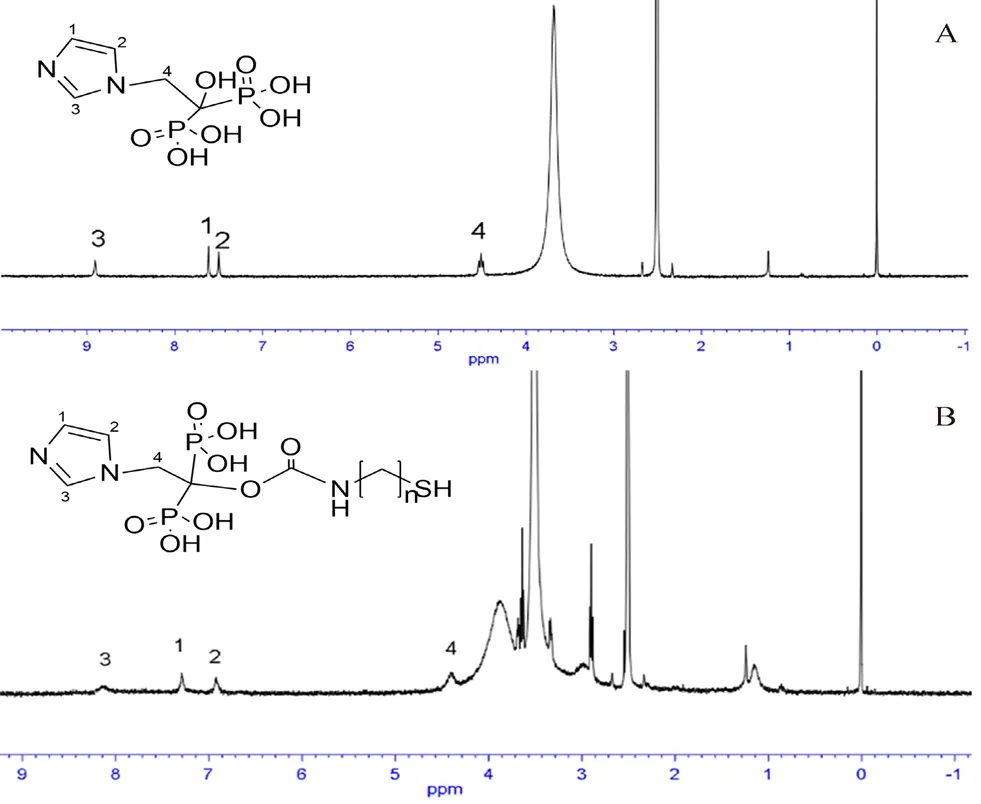
图2 ZOL(A)与ZOL-PEG-SH(B)的1H-NMR 谱图Fig 2 1H-NMR of ZOL(A)and ZOL-PEG-SH(B)
2.3.2 傅里叶变换红外光谱(FT-IR) ZOL与NH2-PEG-SH、ZOL-PEG-SH的红外吸收光谱如图3所示。ZOL-PEG-SH在3143 cm-1、1580 cm-1、1545 cm-1、629 cm-1出现了与ZOL结构中咪唑环化学基团的吸收峰一致的峰。在1635 cm-1出现了吸收峰与酰胺(C=O)的伸缩振动有关的吸收峰,表明形成了新的酰胺键,证实了两药成功合成。MSN和MSN@PDA、MSN@PDA-PEG-ZOL的FT-IR光谱图如图4所示,纳米粒子均在1054 cm-1和965 cm-1处出现吸收峰,分别为Si-O-Si伸缩振动和硅醇基振动。包覆PDA后,在1633 cm-1处的峰归属于芳环骨架的伸缩振动,3385 cm-1处的宽吸收峰归属于N-H/O-H的伸缩振动,证明PDA包覆在MSN表面。而MSN@PDA-PEG-ZOL在1457 cm-1和1349 cm-1处出现的峰表明目标配体ZOL存在于纳米粒子表面上。
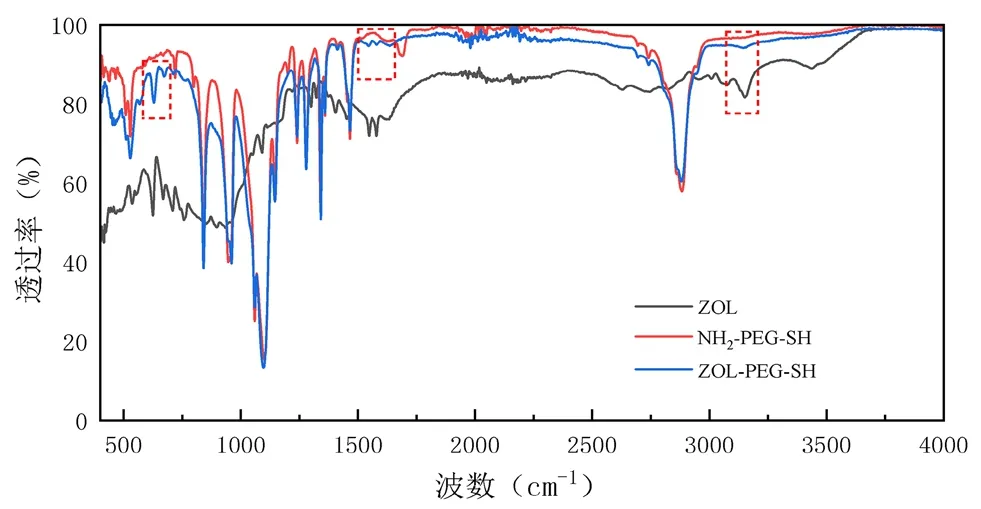
图3 ZOL、NH2-PEG-SH和ZOL-PEG-SH的FT-IR光谱图Fig 3 FT-IR spectra of ZOL,NH2-PEG-SH and ZOL-PEG-SH
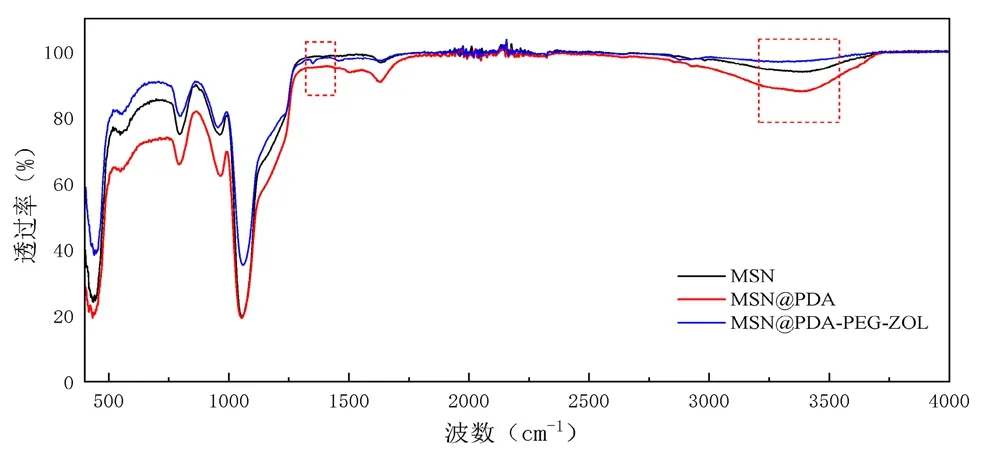
图4 MSN、MSN@PDA和MSN@PDA-PEG-ZOL的FT-IR光谱图Fig 4 FT-IR spectra of MSN,MSN@PDA and MSN@PDA-PEG-ZOL
2.3.3 透射电子显微镜(TEM)分析 取适量纳米粒子进行了TEM分析,观察其纳米粒子形态、尺寸大小。从图5A中可以看到 MSN 表面清晰的介孔结构,其形态为分散均匀的球体,图5B中可以看到PDA包覆后MSN的表面覆盖了一层薄膜,介孔结构被掩盖,表明PDA成功包覆。

图5 MSN(A)和MSN @PDA(B)的TEM照片Fig 5 TEM diagram of MSN(A)and MSN@PDA(B)
2.3.4 马尔文粒度电位仪分析纳米粒子的粒径及Zeta电位 结果如图6所示,MSN的粒径为118 nm,电位为-(24.2±0.4)mV,MSN@PDA粒径为148 nm,电位为-(10.8±0.2)mV,MSN@PDA-PEG-ZOL粒径为(229.1±8.8) nm,电位为-(30.3±0.6)mV,这是由于ZOL在溶液中呈负电性,也说明ZOL成功修饰在纳米粒子表面,纳米粒子的多分散性指数(PDI)在0.21~0.42(在可接受的大小范围内)。

图6 MSN和MSN@PDA、MSN@PDA-PEG-ZOL的粒径(A)和电位(B)Fig 6 Particle size(A)and potential(B)of MSN,MSN@PDA and MSN@PDA-PEG-ZOL
2.3.5 MSN@PDA-PEG-ZOL在水及PBS中的稳定性 连续7 d观察纳米粒子MSN@PDA-PEG-ZOL在37℃的水中及PBS中的稳定性,结果如图7所示,结果表明在两种介质中MSN@3-MA-PDA-PEGZOL均能保持良好的粒径大小,说明纳米颗粒在体内及体外环境下都较稳定。

图7 MSN@PDA-PEG-ZOL在水及PBS中的粒径Fig 7 Particle size of MSN@PDA-PEG-ZOL in water and PBS
2.4 体外药物释放考察
考察MSN@3-MA-PDA与MSN@3-MA-PDAPEG-ZOL在不同pH缓冲液中的释药性。分别取三组样品适量,每组平行三份,每份8 mg,精密称定。于2 mL PBS中超声至完全溶解,放入透析袋(截断分子量3500)。透析袋浸泡在18 mL的PBS(pH为5.0、6.0、7.4)中,调整转速为200 r·min-1。于0.5、1、2、4、6、8、10、12、24、36、48 h 取样 1 mL,同时迅速补加同等体积溶出介质。使用紫外分光光度法测定3-MA含量,并使用公式(3)计算出不同时间累计释放率(CRP)。
式中,Ve为每次所取出的PBS溶液体积(L);V0为缓冲溶液总体积(L);Ci为第i次取出溶液的3-MA浓度(mg·mL-1);n为取液次数;m3-MA为药物载体所负载的3-MA总质量(mg)。
如图8所示,图8B中MSN@3-MA-PDA-PEGZOL在pH 7.4的PBS溶液中释药缓慢,平均释放率仅为(16.99±0.15)%,而在酸性条件下,3-MA的释放度增加,在pH 5.0的PBS溶液中释放率达到(65.11±1.64)%。说明MSN@3-MA-PDA-PEG-ZOL有利于药物在肿瘤微环境中的释放。

图8 MSN@3-MA-PDA(A)与MSN@3-MA-PDA-PEG-ZOL(B)在pH 5.0、6.0、7.4条件下的药物释放曲线Fig 8 Release curves of MSN@3-MA-PDA(A)and MSN@3-MAPDA-PEG-ZOL(B)in PBS(pH 5.0,6.0,and 7.4)
3 讨论
本文使用模板剂法制备了MSN,将PDA包覆MSN纳米粒子后,在其表面修饰ZOL与NH2-PEGSH的连接物,制得具有骨靶向性与pH响应性的载药纳米颗粒MSN@PDA-PEG-ZOL。通过1H-NMR、FT-IR证实ZOL与NH2-PEG-SH连接成功。采用TEM、FT-IR、动态光散射粒径分布仪(DLS)等表征手段对MSN@PDA-PEG-ZOL合成产物进行结构与性能分析。结果表明该纳米颗粒的粒径及Zeta电位分别为(229.1±8.8)nm、-(30.3±0.6)mV;通过浸渍离心法负载3-MA,结果表明,这种多功能MSN纳米颗粒具有高包封率和高载药量,并在体内外具有较好的稳定性。
MSN表面包覆的PDA具有pH响应性,在酸性条件下溶解释放药物,而本试验的体外释药结果也初步证实了在肿瘤微环境中释药量明显高于在正常生理环境下的释药量,这在生物医药领域具有潜在的应用价值。后续将继续深入研究该纳米颗粒在小鼠体内药效学及体内外靶向性,进一步证实纳米粒子对肿瘤的靶向性,为后续骨相关疾病的治疗与自噬抑制剂的递送奠定研究基础。
