Menkes病的临床特点(附1例报告)
2024-03-11王玉孙熙洋王苏悦赵婷婷王训吴君霞赵静年娜许亚运孙丹丹艾文龙付晓明叶群荣李凯张云云
王玉,孙熙洋,王苏悦,赵婷婷,王训,吴君霞,赵静,年娜,许亚运,孙丹丹,艾文龙,付晓明,叶群荣,李凯,张云云
Menkes病(MD)是一种罕见的X-连锁隐性遗传性铜代谢异常疾病[1],国外发病率为1/100000~1/360000,为ATP7A基因(Xq13.2-q13.3)变异所致[2]。因MD具有标志性头发卷曲,故又称“卷发病”。ATP7A基因突变导致肠黏膜铜吸收障碍,铜不能转运至细胞间液及血液循环,导致体内铜缺乏,脑和结缔组织受累,各种铜依赖酶功能障碍,引起以神经系统为主的多脏器损害。MD多于婴幼儿时期起病,主要表现为癫痫、精神运动发育迟滞,肌张力减低。本文报告1例MD,通过总结其临床特点,结合文献进行分析比较,以提高临床医师对本病的认识。
1 临床资料
患儿,男,7月龄,因“发作性全身抽搐、呼之不应4个月”于2022年9月13日入院。患儿约3月龄时出现发作性全身抽搐、呼之不应,表现为双目凝视、上翻,头颈后仰,伴咂嘴样动作,发作频率2~5次/d,发作持续1~20 min/次,抽搐后嗜睡。当地医院考虑“症状性癫痫,脑性瘫痪可能,代谢性脑病可能”,予“丙戊酸钠口服溶液(德巴金)1.5 mL 2次/d、苯巴比妥5 mg 2次/d口服”抗癫痫,无明显改善,此次为进一步治疗就诊于我院。病程中家属称患儿在喂养时反复呛奶,其身高发育轨迹尚正常,但头围、体重偏小,至就诊时仍不能独坐、翻身和爬行,抬头困难。患儿系第2胎第1产,第1胎自然流产,具体情况不详。孕37+2周自然分娩,出生体重3.58 kg,Apgar评分8分,羊水Ⅲ°、量少,脐带绕颈1周,胎盘无异常。患儿出生时因产伤出现顶枕部头皮血肿,并在数小时内进行性增大,于新生儿科对症治疗9 d后头部血肿逐渐消退。患儿出生后反复皮肤黄染,多次光疗退黄后恢复正常。患儿出生后奶粉喂养,按时接种疫苗。患儿父母非近亲婚配,否认癫痫和其他家族遗传性疾病史。入院查体:体重8.5 kg,头围34 cm,体长73 cm。神清,精神一般,反应差,追声、追光不明显,皮肤白皙,小颅畸形,前囟门平软,面颊松弛,鼻扁平,毛发呈黄褐色、分布稀疏,颞枕部毛发略卷曲,高腭弓,胸骨柄下端至剑突局部胸廓下陷、呈漏斗状(图1),心肺听诊(-),腹部平软。双侧瞳孔等大等圆,直径约4.5 mm,直接及间接对光反射灵敏,双侧鼻唇沟对称,四肢肌力检查不配合,粗测双手抓握肌力>Ⅳ°,颈部及四肢肌张力减低,双侧肱二头肌腱、膝腱反射(+),原始病理反射(+)。实验室检查:血常规示白细胞3.55×109/L(参考区间4.8×109/L~14.6×109/L),红细胞3.01×1012/L(参考区间4.0×1012/L~5.5×1012/L),Hb 96 g/L(参考区间97~141 g/L),血小板35×109/L(参考区间190×109/L~579×109/L)。肝功能:丙氨酸氨基转移酶27 U/L,天冬氨酸氨基转移酶60 U/L(参考区间0~50 U/L),白蛋白44.9 g/L,碱性磷酸酶340.0 U/L,胆碱脂酶9676 U/L,余项正常。肌酸激酶540 U/L(参考区间24~194 U/L)。铜蓝蛋白0.04 g/L(参考区间0.2~0.4 g/L),血清铁17.48 μmol/L,血清铜3.12 μmol/L(参考区间10.5~22.0 μmol/L),铜氧化酶0.109 OD(参考区间>0.200 OD),血清锌19.50 μmol/L。肾功能、电解质、C反应蛋白、铁代谢、血氨、乳酸、全段甲状旁腺激素、甲状腺功能五项、CSF生化和常规、CSF涂片抗酸杆菌及细菌、血尿氨基酸筛查均未见明显异常。头颅MRI:T1脑室系统扩张,双侧大、小脑半球脑沟裂明显增宽,胼胝体变薄,T2双侧硬膜下积液,Flair、DWI及磁敏感加权成像(SWI)未见明显异常信号(图2)。腹部彩超:肝胰脾未见明显占位,肝内胆道未见明显扩张,腹腔所见未见明显积液。泌尿系彩超:双侧尿盐结晶。超声心动图检查:房水平少许分流(卵圆孔未闭,直径约1.8 mm)。发作间期EEG:右侧前额、前颞成串中幅不规则慢波同步出现,与左侧不对称。睡眠期左侧枕、后颞单个低-中幅欠典型棘慢波同步出现,可累及左顶区(图3A)。头发光镜下检查:患儿发干内部角质层明显色素减退,中空质脆;外层毛囊边缘可见鳞状突起;毛发断端粗糙杂乱,似“扫帚”或“毛刷”样(图4)。家属知情同意后,抽取患儿及其父母的抗凝外周血,委托北京全谱医学检验实验室行全外显子组检测及线粒体DNA全测序。结果显示,在该患儿ATP7A基因(NM_000052)检出1个剪接位点变异,即c.2916+2(IVS14)T>C(图5)。该位点的美国医学遗传学与基因组学学会(ACMG)临床致病性证据为可能致病的。但该位点在千人基因组、gnomAD、gnomAD东亚人群数据库未见该突变频率报道,且未被ClinVar数据库收录。Sanger测序验证示患儿母亲为表型正常的杂合携带者,该变异来源于患儿母亲,而患儿父亲为正常野生型。另外,拷贝数变异和线粒体基因结果均为阴性。诊断:MD。本病无根治手段,调整抗癫痫药物为德巴金2 mL 2次/d口服,奥卡西平口服混悬液(曲莱)2 mL 2次/d口服,患儿癫痫发作次数减少,发作持续数秒,表现为双上肢不对称性伸展、上抬,点头样动作,偶有右上肢快速抽动。复查EEG清醒安静状态下持续弥漫性不规则慢波,其中夹杂大量不同步、不对称的高-极高幅棘慢、尖慢、多棘慢波;背景呈不典型“高度失律”改变(图3B)。由于患儿脑电较前恶化,在与患儿家属充分沟通病情后,建议至上级儿童医院行对症和支持治疗。

图1 患儿皮肤、毛发及外观A:皮肤白皙,漏斗胸;B:面颊松弛,鼻扁平,毛发呈黄褐色、分布稀疏;C:颞枕部毛发略卷曲;D:高腭弓畸形。
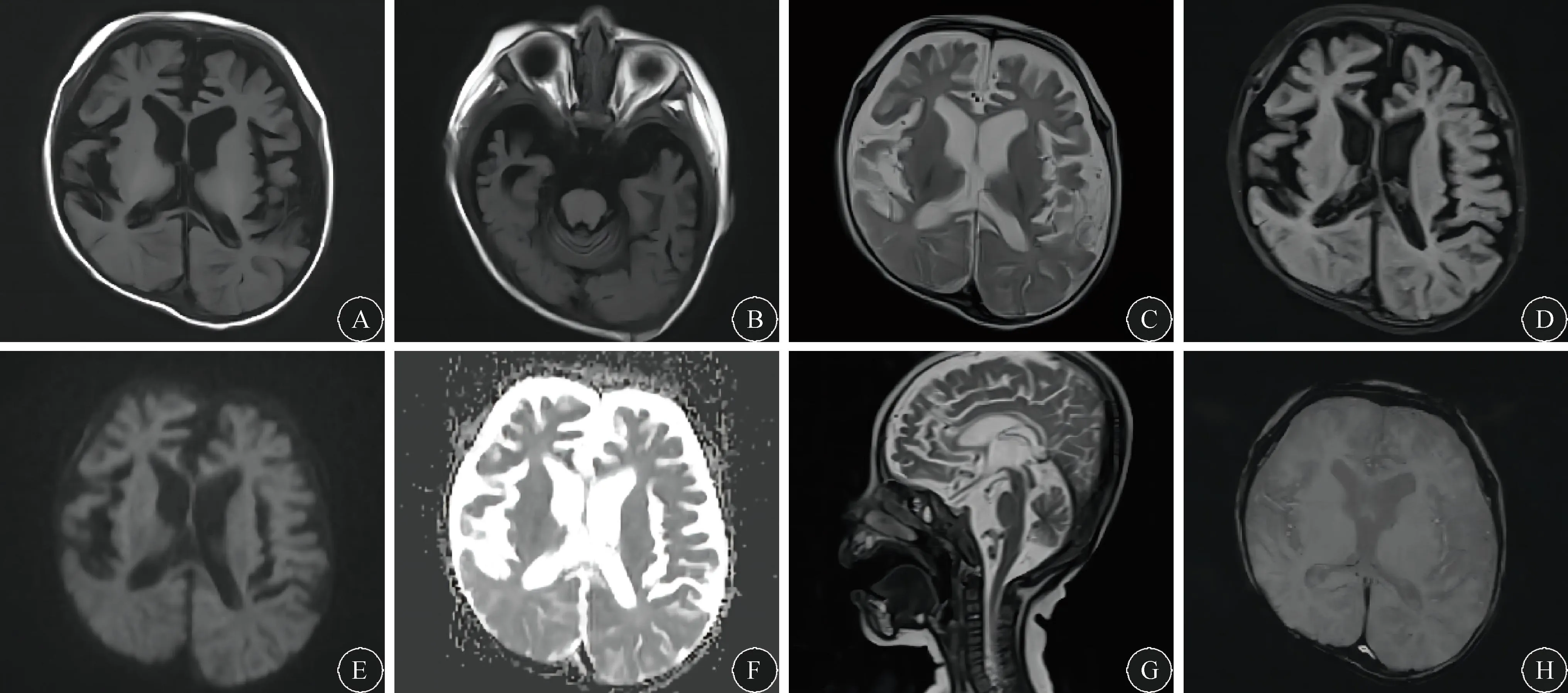
图2 患儿头颅MRI检查 A、B:轴位T1显示大、小脑半球沟裂增多、增宽,脑室系统扩张,胼胝体发育不良;C:轴位T2显示双侧硬膜下积液;D、E、F:轴位Flair相、DWI及表观弥散系数未见明显异常信号;G:矢状位T2可见胼胝体变薄、小脑明显萎缩;F:轴位SWI相未见含铁血黄素沉积。

图3 发作间期EEG A(2022-9-22):全头部广泛性40~120 μV、1.2~2 Hz多形性δ波,未成调节调幅,双侧基本对称。各导间可见较多低-中幅5~7 Hz θ波及低幅β波。睁眼时α节律可见部分抑制。右侧前额、额、前颞成串中幅2~4 Hz不规则慢波同步出现,与左侧不对称。B(2022-10-6):持续弥漫性不规则200~250 μV,1.5~2.5 Hz不规则δ波,兼见较多中-极高幅4~6 Hz θ波活动波及15~20 Hz低幅β波,其中大量不同步、不对称的高-极高幅棘慢、尖慢、多棘慢波夹杂。背景呈不典型“高度失律”改变。

图4 光镜下患儿毛发A:左侧为患儿毛发,可见发干内部角质层明显色素减退,中空质脆,外层毛囊的边缘可见鳞状突起,右侧为健康7月龄男婴毛发(×200);B:患儿毛发的断端粗糙杂乱,似“扫帚”或“毛刷”样(×400)。

图5 患儿及其父母测序峰图A:先证者(Ⅱ-1)为ATP7A基因半合子,检出剪接位点变异[NM_000052:c.2916+2(IVS14)T>C];B:患儿父亲(Ⅰ-1)为表型正常的野生型;C:患儿母亲(Ⅰ-2)为表型正常的ATP7A基因杂合子。
2 讨 论
MD是一种以进行性神经功能恶化、结缔组织病和异常毛发为临床特征的先天性铜吸收、分布障碍性疾病[3]。MD基本只在男性患儿中发病,女性多为携带者,由定位于X染色体Xq13.2-q13.3上的ATP7A基因突变导致。该基因负责编码P型跨膜铜转运ATP酶A型。ATP7A基因突变可致肠道无法正常吸收食物中的铜,同时阻碍铜的跨血-脑屏障转运,使包括CNS在内的全身组织、器官发生铜吸收和分布障碍,机体处于铜缺乏状态;另外,继铜缺乏后又会导致参与线粒体能量代谢、铁稳态、黑色素合成、神经传递和结缔组织发育的多种铜依赖酶(分别为细胞色素c氧化酶、铜氧化酶/铁氧化酶、酪氨酸酶、多巴胺β-羟基酶和赖氨酸氧化酶)活性降低,从而发生相应的铜缺乏症状[4]。MD具有一定的表型变异性和异质性,本例患儿为经典型MD,发病早、表型重、预后差,多于2~3月龄起病,3岁前死亡。另两种相对较轻、发病延迟的ATP7A相关铜转运障碍性疾病分别为枕角综合征(OMIM 304150)和ATP7A相关远端运动神经病变(OMIM 300489),这两种ATP7A蛋白影响较轻的亚型暂不赘述。
癫痫是MD的重要临床表现,但因可供研究的病例数较少,过去零星的报道无法反映MD癫痫临床全貌,其病理生理机制亦不完全清楚,可能涉及多种铜依赖酶活性受损,或与铜代谢紊乱及其继发的脑结构异常有关;而且铜作为抗N-甲基-D-天门冬氨酸(NMDA)受体的非竞争性拮抗剂,在ATP7A蛋白活性缺乏的情况下,会加剧海马神经元NMDA受体介导的兴奋毒性,产生级联反应[5]。Bahi-Buisson等[6]通过回顾性分析12例癫痫MD患儿,首次将MD癫痫分为三个阶段,并总结其EEG特征,分别为:(1)早期<3个月内,以局灶性发作(强直-阵挛发作、阵挛发作)为特征,通常由发热诱发发作,常演变为癫痫持续状态。早期发作期的EEG表现为后头部成串棘慢波、慢波,发作间期EEG呈多灶性、多形性棘慢复合波。(2)中期在6~11个月内,以婴儿痉挛症为特征。中期发作间期EEG特征为改良型高度失律或弥漫性、非对称棘慢波。(3)晚期在20~25个月内,以多灶性癫痫发作、姿势强直性痉挛和肌阵挛发作为特征。晚期发作间期EEG均显示多灶性高振幅度活动,伴不规则慢波。Verrotti等[7]报道了28例癫痫MD患儿的多中心、电-临床研究,是迄今规模最大的长期随访研究,结果在这些MD患儿中并未发现典型3阶段性癫痫进程,其中最常见的癫痫发作类型是局灶性发作和婴儿痉挛症,EEG特征是高度失律及间歇期的节律慢化。本例患儿在3月龄时以局灶起源的全面性阵挛发作起病,伴口腔自动症,先后予3种抗癫痫药后仍控制不佳,7月龄癫痫发作类型渐演变为强直发作、痉挛发作,偶伴肌阵挛发作,EEG背景呈不典型“高度失律”。因此,结合本例情况,考虑以婴儿痉挛症为代表的多种类型癫痫发作是MD癫痫的临床特点,这与Gkampeta等[8]的观点相一致,若临床上发现具有皮肤、毛发特殊改变的婴儿痉挛症或难治性癫痫患儿[9],在遗传性病因中需要重点考虑MD[10]。
毛发异常是MD的代表性体征,不同于自然卷发。铜缺乏导致酪氨酸酶生成不足,而酪氨酸酶与头发、皮肤色素含量有关,因此会造成患儿头发和皮肤色淡;赖氨酸氧化酶活性降低会显著减少结缔组织的强度,表现为毛发短粗卷曲,质脆易断,形似钢丝清洁球,以颞部、枕后部为著。借助光镜可以更清楚地观察到毛发色素减退、发干扭曲和断裂,其镜下特点主要为:(1)局限性狭窄造成的串珠样改变;(2)横向扭曲发生的断裂及脆发结节;(3)纵向撕裂形成的羽状脆发。肉眼可见本例毛发分布以颞、枕部较稀疏,略卷曲,形态细软,在中倍光镜下可观察到明显的色素减退,高倍镜下可以观察头发断端粗糙杂乱,符合MD的毛发特征。由于毛发发育异常是多种遗传性疾病(如Griscelli综合征、Chediak Higashi综合征)的重要诊断线索之一,有学者[11]建议采用更高效清晰的皮肤镜检,以提高检出率,和减少取样毛发的附带伤害。但实际工作中凭毛发外观早期识别MD并不容易,因为患儿在新生儿期的毛发外观可正常,当新生儿脱发后,可能才会出现典型的“卷毛”或“钢发”。
过去诊断MD除了依据特征性临床表现外,还需要铜蓝蛋白、血清铜减低作为铜代谢异常的佐证。本例铜蓝蛋白、血清铜明显减低,符合传统意义上的MD生化特征。但需注意的是,因血清铜蓝蛋白浓度在不同年龄阶段波动较大[12],新生儿时期约为成人1/4~1/5,至1岁左右达正常成人水平,健康新生儿及婴幼儿的铜蓝蛋白可呈生理性减低,造成假阳性结果,故铜蓝蛋白减低并不一定代表铜缺乏;另外,铜蓝蛋白作为一种急性时相反应蛋白,在感染、炎症或创伤时也会短暂地升高,造成假阴性结果。检测患儿铜蓝蛋白的实际临床意义有限,故不能仅凭铜蓝蛋白减低诊断该年龄阶段的MD,建议可同时检测铜氧化酶活性间接反映真实的铜蓝蛋白水平。此外,肝豆状核变性(OMIM 277900)为常染色体隐性遗传的铜排泄障碍病,以体内铜过剩为特征,无铜蓝蛋白血症(OMIM 604290)为常染色体隐性遗传的铁沉积性疾病,这两种疾病与MD同为金属代谢障碍病,而且致病基因具有一定同源性、临床表现相近、均以铜蓝蛋白减低为生化特点[13],临床上需注意鉴别(表1)。因铜缺乏会降低多巴胺β-羟基酶的活性,导致血浆中多巴胺(DA)升高和去甲肾上腺素(NE)减低。Goldstein等[14]研究发现,血浆DA/NE比值(>0.2)及其代谢产物二羟基苯乙酸/二羟基苯乙二醇比值(>5)对新生儿MD的诊断具较高的敏感性和特异性。巴西MD循证指南[15]也推荐首选此法诊断新生儿MD。但由于神经递质检查对标本采集、运输和实验室技术要求较高,目前我国对于疑诊MD的患儿并不常规检测儿茶酚胺中的DA/NE比值。
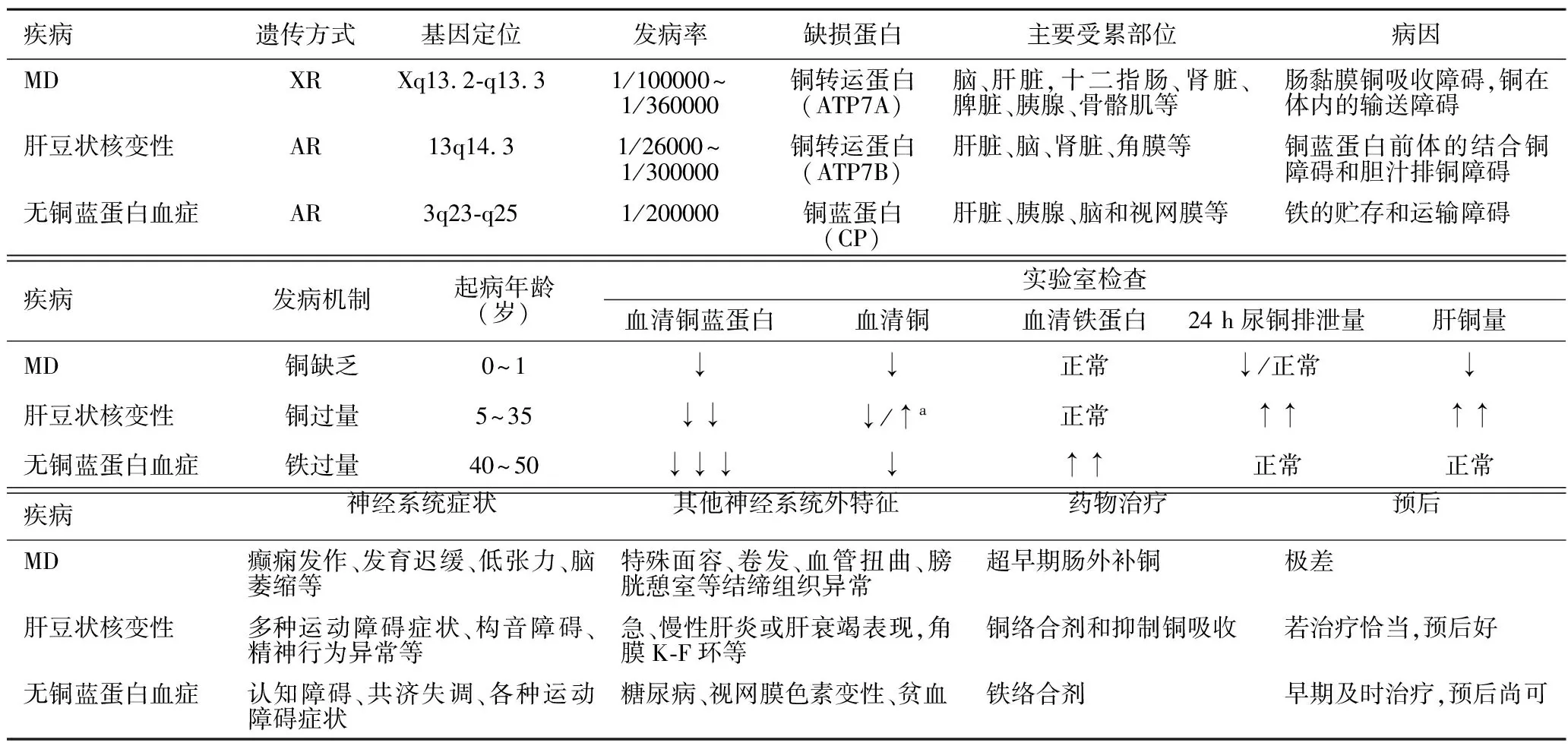
表1 Menkes病、肝豆状核变性与无铜蓝蛋白血症的鉴别诊断
有多中心、影像学回顾性研究[16-17]表明,MD的头颅MRI特征主要包括:(1)白质异常,包括先天性的髓鞘发育不良、可能与铜依赖酶参与的代谢障碍相关的脑白质瘤样病变与局灶性非瘤样白质病变;(2)进行性弥漫性脑萎缩,尤其是小脑萎缩;(3)基底神经节受损;(4)硬膜下积液或血肿,可能与血管壁脆性增加有关;(5)颅内血管异常,MRA和MRV显示颅内“螺丝锥”样血管迂曲延长是最为突出的表现之一,具有诊断学意义,其原因与赖氨酰氧化酶参与弹性蛋白和胶原的交联作用下降导致的血管壁结构损伤有关。SWI则有助于发现含铁血黄素的沉积,判断有无因结缔组织脆性增加造成的颅内出血。本例患儿头颅MRI示弥漫性脑萎缩,皮、髓质脑发育不良,以额、颞叶和小脑最为明显;双侧额、顶叶可见硬膜下积液。不足的是,由于患儿在院期间洗澡受凉后出现发热、哭闹明显、不愿进食,未能配合完成MRA检查。
ATPTA基因突变的外显性达100%,基因是诊断MD的金标准,诊断通常建立在男性先证者检出ATP7A半合子致病性变异的基础上。ATP7A基因全长约150 kb,包括23个外显子。已发现该基因有三百多种变异位点,包括小的插入和缺失(35%)、无义突变(20%)、剪接(15%)和错义突变(8%)以及大片段的缺失或重排(20%)等,其中1/3是新发突变。该基因虽然与肝豆状核变性的基因(ATP7B)相似,二者有56%的同源性,但作用不同。前者在十二指肠、小肠上部、肾、脑、心脏、肺、肌肉、胰及胎盘细胞中均有表达,但在肝脏没有。
关于MD的治疗,目前尚无根治手段,对症支持治疗仍是主要方法。虽然肠外补铜已被证实是安全有效的疾病修饰疗法,若能在出生后30 d内接受组铵酸铜皮下注射,可一定程度预防和延缓病情进展,但是补铜无法逆转已发生的神经功能障碍,对于已经发生癫痫的MD患儿无法改善长期预后和最终结局[7]。国内尚无组氨酸铜渠道来源,治疗仍限于抗癫痫、营养支持、康复训练等。
本例病例报道的临床表型符合经典型MD特征,其癫痫和脑电类型、面容和毛发特征、铜生化和头颅MRI改变与既往报道病例一致,行基因检测发现了新的致病性变异c.2916+2(IVS14)T>C,扩大了MD基因的突变谱,为MD的分子诊断增加了新的临床依据。至于ATP7A基因与表型的相关性,未来可能还需要更多临床资料与更长周期的临床观察加以探索。
作者贡献说明王玉、孙熙洋、王苏悦提出选题,设计、实施研究,采集、分析、解释数据,撰写文章;赵婷婷、年娜、许亚运、孙丹丹、艾文龙、付晓明指导选题,指导起草,提供研究经费;王训、吴君霞、赵静、叶群荣、李凯、张云云指导文章修改,提供技术材料
