Predicting colorectal cancer prognosis based on long noncoding RNAs of disulfidptosis genes
2024-03-08KuiLingWangKaiDiChenWenWenTangZePengChenYuJiWangGuoPingShiYuGenChen
Kui-Ling Wang,Kai-Di Chen,Wen-Wen Tang,Ze-Peng Chen,Yu-Ji Wang,Guo-Ping Shi,Yu-Gen Chen
Abstract BACKGROUND A recently hypothesized cause of cell death called disulfidptosis has been linked to the expansion,emigration,and vascular rebuilding of cancer cells.Cancer can be treated by targeting the pathways that trigger cell death.AIM To discover the long non-coding RNA of the disulfidaptosis-related lncRNAs (DRLs),prognosis clinical survival,and treat patients with colorectal cancer with medications.METHODS Initially,we queried the Cancer Genome Atlas database to collect transcriptome,clinical,and genetic mutation data for colorectal cancer (CRC).Training and testing sets for CRC patient transcriptome data were generated randomly.Key long non-coding RNAs (lncRNAs) related to DRLs were then identified and evaluated using a least absolute shrinkage and selection operator procedure,as well as univariate and multivariate Cox regression models.A prognostic model was then created after risk scoring.Also,Immune infiltration analysis,immune checkpoint analysis,and medication susceptibility analysis were used to investigate the causes of the different prognoses between high and low risk groups.Finally,we validated the differential expression and biomarker potential of riskpredictive lncRNAs through induction using both NCM460 and HT-29 cell lines,as well as a disulfidptosis model.RESULTS In this work,eight significant lncRNAs linked to disulfidptosis were found.Gene ontology and Kyoto Encyclopedia of Genes and Genomes pathway enrichment analyses of differentially expressed genes between high-and low-risk groups from the prognostic model showed a close relationship with the immune response as well as significant enrichment in neutrophil extracellular trap formation and the IL-17 signaling pathway.Furthermore,significant immune cell variations between the high-risk and low-risk groups were seen,as well as a higher incidence of immunological escape risk in the high-risk group.Finally,Epirubicin,bortezomib,teniposide,and BMS-754807 were shown to have the lowest sensitivity among the four immunotherapy drugs.CONCLUSION Our findings emphasizes the role of disulfidptosis in regulating tumor development,therapeutic response,and patient survival in CRC patients.For the clinical treatment of CRC,these important LncRNAs could serve as viable therapeutic targets.
Key Words: Colorectal cancer;Clinical outcomes;Disulfidptosis;Drug sensitivity;Immunotherapy
lNTRODUCTlON
Colorectal cancer (CRC),the third most prevalent cancer in the world,accounting for over 1.8 million new cases and 881000 fatalities per year[1].Via the use of auxiliary diagnostic procedures such as biochemical testing,digital rectal examination,sigmoidoscopy,and colonoscopy,the detection and survival rates of CRC have gradually improved[2,3].However,among people under 50,the incidence and metastatic rates have been steadily increasing[4,5],with the incidence of CRC among adults under 50 years old increasing annually between 2012 to 2016 at a rate of 2.2%[6].Nevertheless,CRC is also a heterogeneous disease[7],and challenges like microsatellite instability and chromosomal instability brought on by gene mutations,as well as multidrug resistance caused by tumor heterogeneity can affect treatment outcomes[8].Thus,to choose the best chemotherapeutic treatments,identify sensitive groups,diagnose early stages of the disease,and increase the efficacy of diagnosis and treatment,it is desirable to investigate new biomarkers[9].
Recently,a process of cellular death known as,which is associated with the buildup of intracellular disulfide compounds,was postulated.This may therefore signify a change in how tumors are treated.Solute Carrier Family 7 Member 11 (SLC7A11) is overexpressed in the tumor microenvironment,where it speeds up the transport of cysteine into cells.In addition,injured immune cells and tumor cells have high metabolic rates and eat large amounts of glucose,which therefore causes a shortage of the reducing agent nicotinamide adenine dinucleotide phosphate (NADPH) and triggers glucose deprivation[10].The reduction of cysteine to cystine is impacted by enhanced cysteine transport and the absence of NADPH,and in turn causes an accumulation of intracellular disulfide compounds.This accumulation then causes cell death following alteration of the cytoskeletal protein shape[11].When cells overexpressing SLC7A11 are starved of glucose,Liuetal[12] found that supplementing culture media with 2-mercaptoethanol—a chemical that breaks disulfide bonds—can prevent defects induced by oxidative stress and therefore prevent cell death.Overall,disulfidptosis is a novel target for tumor therapy because it disturbs the integrity of the tumor microenvironment[13].
Noncoding RNAs longer than 200 nucleotides are known as long noncoding RNAs (lncRNAs).They are known to control the proliferation,differentiation,invasion,and metastasis of cancer cells by interacting with DNA,RNA,proteins,or lipids[14,15].In addition,lncRNAs can control how metabolically relevant proteins undergo post-translational changes and support cancer energy metabolism[16].Moreover,it has been widely documented that lncRNAs and CRC have a regulatory link.LncRNAs function as signaling molecules in pathways relevant to CRC or as competitive endogenous RNAs (ceRNAs) by competitively binding to common microRNA (miRNA) binding sites.This sequesters miRNAs and changes the expression of downstream target genes[17].For example,by controlling the focal adhesion signaling pathway,the lncRNA ITGB8-AS1 functions as a ceRNA to enhance CRC cell proliferation and tumor formation[18].Moreover,CRC can be efficiently inhibited by targeting ITGB8-AS1.In addition,studies have shown that lncRNAs play a role in many stages of CRC,including intestinal polyps and distant metastases,making them attractive prospective targets for treatment[19].
At present,there is no published data regarding the regulatory connection between disulfidaptosis-related lncRNAs (DRLs) and CRC.Consequently,the objective of this study is to determine whether DRLs can be used as a tumor biomarker for CRC to forecast patients prognoses and responses to treatment.Our study therefore offers a novel approach for forecasting how cancer patients respond to treatment and how they may fare clinically.
MATERlALS AND METHODS
Data sources
First,we obtained transcriptomic data for the Cancer Genome Atlas (TCGA)-COAD and TCGA-READ,including 564 CRC tumor samples and 44 normal tissue samples.Next,we ran Perl version 5.30.0 to extract RNA information from transcriptomic data and matched them with clinical datasets containing variables such as sex,age,stage,and survival time[20].
Screening for lncRNAs co-expressed with disulfidptosis genes
First,we searched for “disulfidptosis” in PubMed (https://pubmed.ncbi.nlm.nih.gov/?db=pubmed) and identified several genes connected to this molecular function.Next,we used the R packages “BiocManager” and “limma” as implemented in R version 4.2.0[21] to obtain an LncRNA expression matrix for disulfidptosis.|Pearson R| > 0.5 andP< 0.001 were used as filter conditions to produce an expression matrix for DRLs.We then used the “ggplot2” and “ggalluvial” R packages to create a Sankey association between genes related to disulfidptosis and DRLs[22].
Building and validating a risk prognosis model
Next,we combined clinical survival data with the LncRNA expression matrix,and omitted patients with missing information.CRC patients were then randomly allocated to training and testing groups.The testing group and the full dataset were used to validate the accuracy of the prognostic model,while the training group was used to construct the prognostic model.Next,to test the DRLs of the prognostic model,we used least absolute shrinkage and selection operator (LASSO) and univariate Cox regression analysis,followed by multivariate Cox regression analysis,at a significance threshold ofP< 0.05.Eight DRLs were obtained.Relevant risk curves and heatmaps were then plotted using the “survival,” “glmnet,” “survminer,” and “timeROC” R packages[23,24].The following equation was then used to obtain the risk score[25]: Risk score=Σi=lnCoef (i) × Expr (i).The risk expression (Expr(i)) and risk coefficient (Coef(i)) functions represent the corresponding risk coefficient and risk expression.The samples in the training and testing groups were split into high-risk and low-risk groups based on the median values of the risk ratings assigned to each patient in the training group.The R packages “survival,” “replot,” and “rms” were used to create receiver operating characteristic (ROC) curves,c-index plots,column line plots,and calibration plots to evaluate the independence and accuracy of the prognostic model[26].
Gene ontology, Kyoto encyclopedia of genes and genomes, and gene set enrichment analysis pathway enrichment analysis
Next,we performed gene ontology (GO) functional enrichment analysis,Kyoto encyclopedia of genes and genomes (KEGG) pathway enrichment analysis,and gene set enrichment analysis (GSEA) enrichment analysis on the identified differentially expressed genes (DEGs) in the high-risk and low-risk groups,respectively.These analyses used aPvalue cutoff of 0.05 and a q-value cutoff of 0.05,and was implemented using the R software packages colorspace,stringi,DOSE,clusterProfiler,and enrichplot.
Tumor mutational burden and immune dysfunction and exclusion analyses
The tumor mutational burden (TMB) for each patient was determined using Perl scripts to analyze CRC mutation data.We carried out differential analysis using the R software packages “ggpubr,” “limma,” “survival,” and “survminer.” The “ggpubr” and “limma” packages.Next,the tumor immune dysfunction and exclusion (TIDE) score file was acquired from the TIDE website (http://tide.dfci.harvard.edu),and violin plots were created to compare the immune escape capabilities of high-and low-risk groups with respect to CRC TIDE[27].
Analysis of differences in the tumor microenvironment
We then used the “reshape2” and “ggpubr” packages to compare the immunological characteristics of the high-and lowrisk cluster groups,and used box plots to illustrate the abundances of 22 tumor-infiltrating immune cells.We also used the “limma,” “reshape2,” “tidyverse,” “ggplot2,” “ggpubr,” and “ggExtra” R packages to create a correlation heatmap for risk score,immune cell infiltration,and immune checkpoint analysis.
Analyses of drug susceptibility
The Genomics of Drug Sensitivity in Cancer (GDSC) website (https://www.cancerrxgene.org) was used to extract drug sensitivity and expression data for targeted therapies.OncoPredict software was used to forecast how responsive patients in the high-and low-risk groups would be to therapeutic medications[28,29].
Cell lines and cell culture
Human colorectal adenocarcinoma cells (HT-29) and normal colonic epithelial cells (NCM460) were procured from the Cell Repository at the Shanghai Institute of Cell Research (Shanghai,China).Both cell lines were authenticated using short tandem repeat analysis,and mycoplasma testing results were negative.NCM460 cells were cultured in RPMI-1640 medium (Gibco,California,United States),while HT29 cells were grown in Dulbecco's Modified Eagle Medium (DMEM) (Gibco,United States).Both culture media were supplemented with 10% fetal bovine serum (FBS,Sangon Biotech,China),as well as 100 U/mL penicillin and streptomycin (PS,Gibco,Shanghai,China).Additionally,both cell lines were maintained in a humidified incubator at 37 °C with 5% CO2.
Disulfidoptosis cell model establishment and cell proliferation assay
The experimental procedure was based on previously established protocols.2-fluoro-6-(m-hydroxybenzoyloxy) phenyl m-hydroxybenzoate (WZB117) is a glucose transporter 1 (Glut1) inhibitor,effectively suppressing glucose uptake.In cancer cells with overexpression or underexpression of SLC7A11,WZB117 treatment creates a glucose-deficient intracellular environment,significantly reducing the production of NADPH in the pentose phosphate pathway.Insufficient NADPH impairs the ability to reduce accumulated intracellular cysteine,inducing disulfide stress and leading to cell death[12].Therefore,WZB117 can mimic the intracellular conditions of glucose starvation,making it an inducer of disulfidoptosis.
Cell proliferation was assessed using the cell counting kit-8 (CCK8) assay kit (Bio-Rad Laboratories,Inc.) according to the manufacturer's instructions[30].HT-29 cells were seeded at a density of 5-6 × 103cells per well in a 96-well plate and cultured at 37°C for 24 h.After removing the culture medium,different concentrations of WZB117 (0,1,3,10,15,30,50,100,300 µmol/L) were added,and the cells were incubated for an additional 24 h.Subsequently,the culture medium was removed,and 100 µL of basal culture medium and 10 µL of CCK8 reagent were added to each well.The cells were then cultured for 4 h,and the absorbance at 450 nanometers was measured to assess cell viability under various drug concentrations.
Quantitative real-time polymerase chain reaction analysis
NCM460 cells and HT29 cells were seeded in 6-well plates (1-2 × 105cells per well).HT-29 cells were treated with WZB117 (0 and 300 μmol/L) for 24 h.After 24 h,the culture medium was removed,and cells were washed 2-3 times with PBS at 4°C,harvested,and lysed.Total RNA from NCM460 and HT-29 cells was extracted using the RNA isolator (Vazyme,Nanjing,China).Complementary DNA (cDNA) was synthesized from 1 ng of total RNA using the NovoScript Plus All-in-one 1st Strand cDNA Synthesis SuperMix Kit,which contains genomic DNA (gDNA) removal (Jiangsu Novogene Bioinformatics Technology Co.,Ltd.).Real-time quantitative polymerase chain reaction (qPCR) was performed using the QuantStudio 5 Real-Time PCR System (Applied Biosystems,United States) with the Hieff qPCR SYBR Green Master Mix (Shanghai Yisen Biological Technology Co.,Ltd.).The primers listed in Table 1 were synthesized by Sangon Biotechnology (Shanghai) Co.,Ltd.
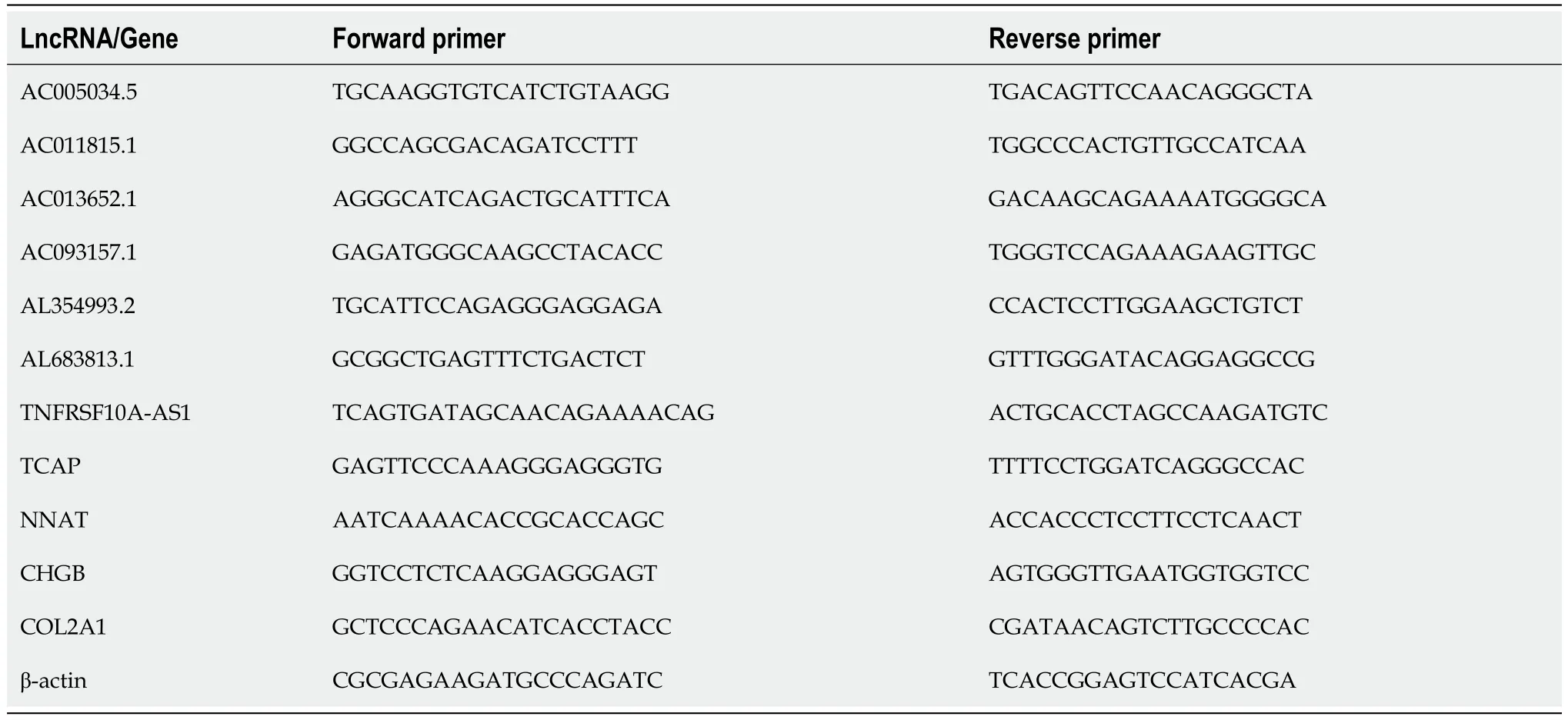
Table 1 Primers for disulfidptosis-related lncRNAs/Genes
RESULTS
Expression of LncRNA associated with co-expression of the disulfidptosis gene in CRC
A total of 571 CRC expression sequences and normal sample expression sequences were discovered using the TCGA transcriptome data.In addition,a literature search identified ten disulfidptosis genes (Supplementary Table 1).We then identified 564 differentially regulated lncRNAs following a correlation analysis between the disulfidptosis genes and CRC LncRNA dataset (|Pearson’s R| > 0.5 andP< 0.001;Supplementary Table 2).Subsequent patients with incomplete data were then eliminated and the CRC LncRNA expression matrix was combined with clinical survival data,so we then obtained a total of 542 CRC patients in an expression matrix who satisfied all criteria.We found no significant variation in the clinical characteristics of the two groups between the 542 CRC patients randomly between the training group (n=271) and the testing group (n=271;Table 2).The relationship between DRLs and disulfide death genes was depicted using a Sankey diagram (Figure 1A).Next,using LASSO and univariate Cox regression analyses,12 DRLs associated with CRC prognosis were identified,including five protective DRLs (i.e.,SNHG16,AC093157.1,AC005034.5,TNFRSF10A-AS1,and AC011815.1) and seven DRLs with a hazard ratio (HR) > 1,which indicate adverse prognostic factors (Figure 1B-D).Finally,eight DRLs were discovered that were related to overall survival (OS) in the TCGA-COAD and TCGA-READ cohorts based on multivariate Cox regression analysis (Supplementary Table 3).A correlation heatmap was then created to show the co-expression patterns of these eight DRLs (i.e.,AC005034.5,AC006213.7,AC011815.1,AC013652.1,AC093157.1,AL354993.2,AL683813.1,and TNFRSF10A-AS1),which were thought to have the most significant impact on CRC prognosis (Figure 1E).
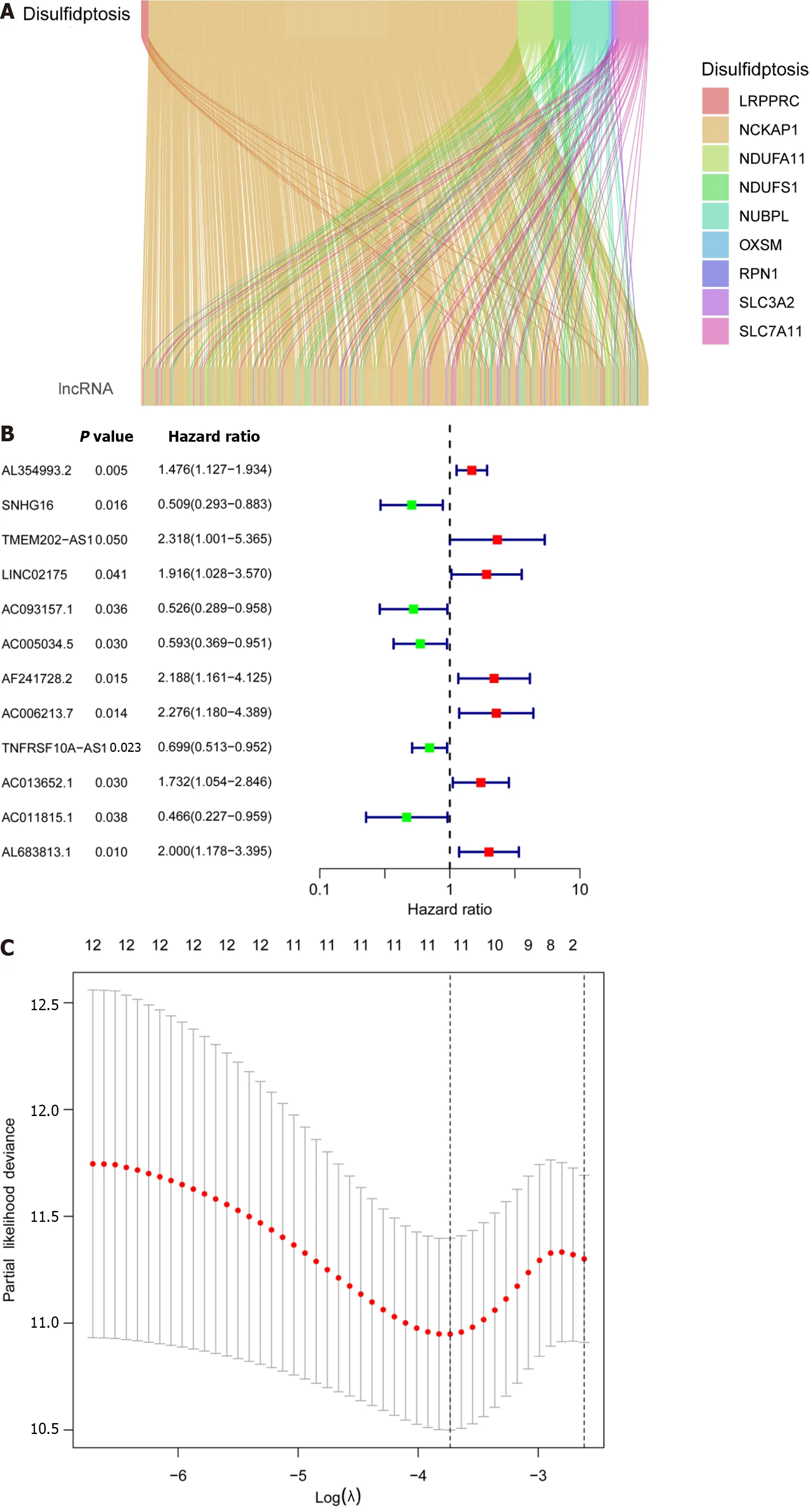
Figure 1 Prognostic features of disulfidaptosis-related lncRNAs in colorectal cancer. A: Sankey diagram showing the relationship between disulfidaptosis-related genes and disulfidaptosis-related lncRNAs (DRLs);B: Forest plot of prognostic genes associated with DRLs;C: The least absolute shrinkage and selection operator (LASSO) coefficients of DRLs obtained via LASSO analysis;D: Cross-validation of DRLs in LASSO regression;E: Multivariable Cox regression analysis and correlation between disulfidaptosis genes and DRLs.aP < 0.05;bP < 0.01;cP < 0.001.
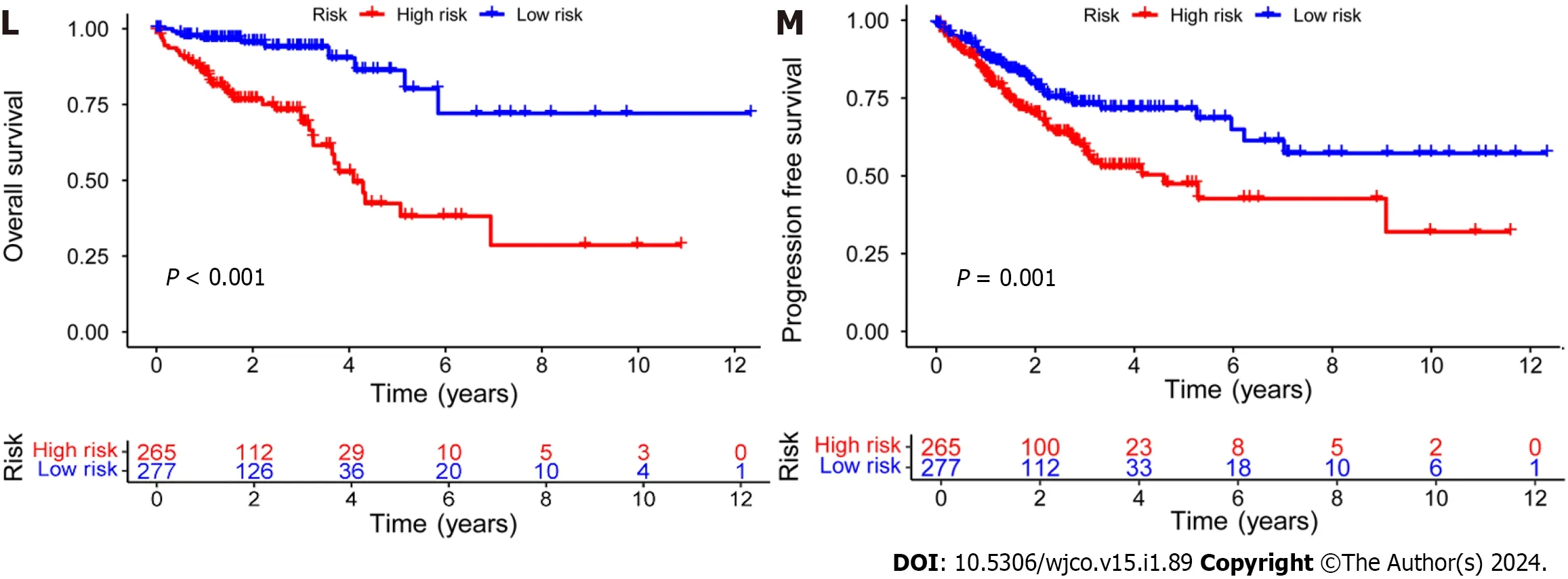
Figure 2 Establishment of a risk prediction model for predicting overall survival in colorectal cancer patients. A-C: Distribution of patients in the training,testing,and combined sets with increasing risk scores;D-F: Survival time and risk scores of patients in the three groups;G-I: Risk score heatmaps for eight key disulfidaptosis-related lncRNAs in the three groups;J-L: Kaplan-Meier survival analysis of overall survival in the three groups of colorectal cancer patients;M: Progression-free survival analysis of the combined dataset.
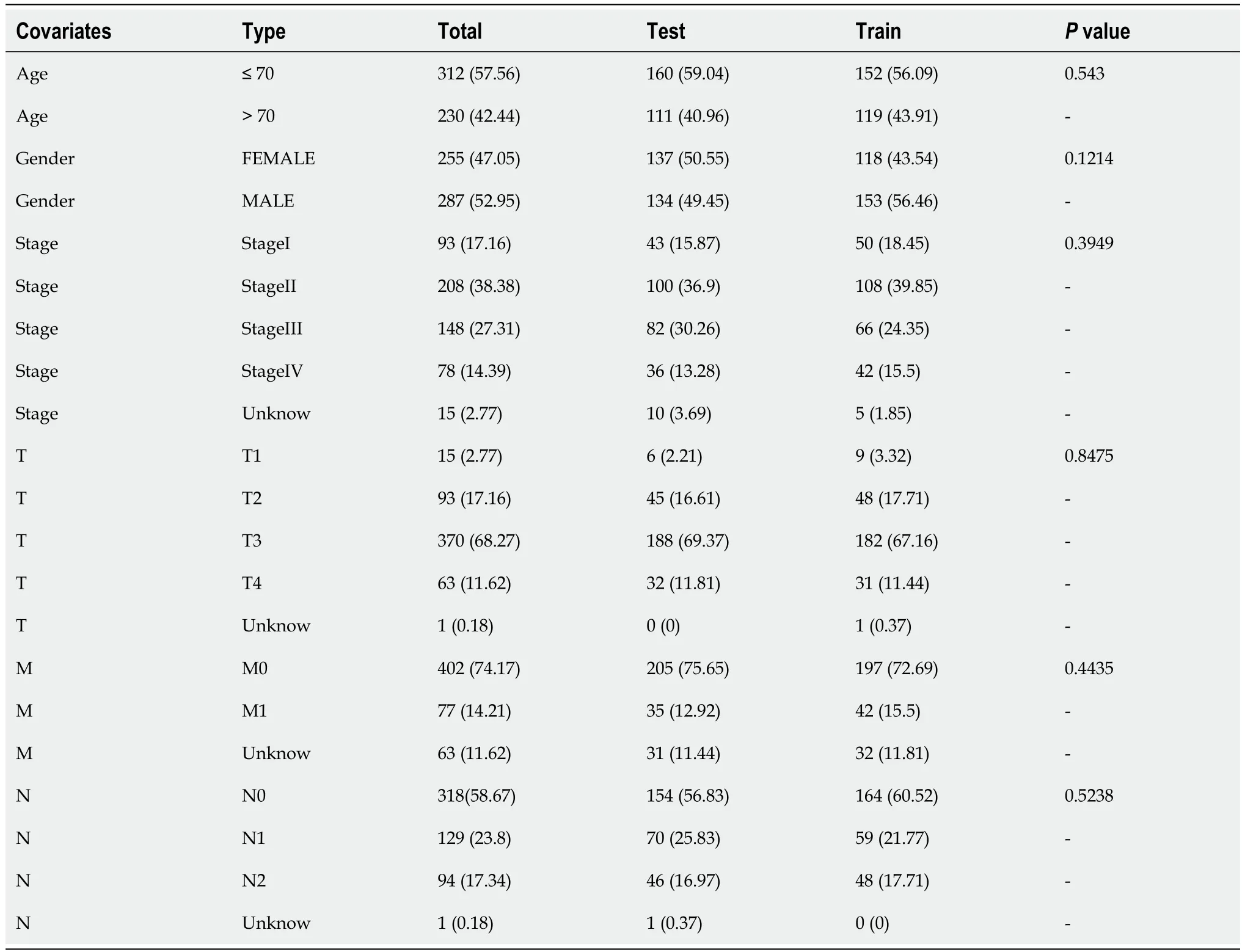
Table 2 Clinical characteristics of colorectal cancer patients in the training and testing groups,n (%)
Building a risk prediction model to predict OS in CRC patients
Subsequently,the risk score formula was utilized to calculate each score to generate the risk curve.The patient sample,ranked from low risk to high risk,is the abscissa in Figure 2A-C,while the ordinate represents the risk score value.542 CRC patients were categorized as high risk or low risk based on the training group risk score's median risk score value.The survival time of the patients in the training group,test group,and all sets were subsequently analyzed (Figure 2D-F).That was discovered that the number of dead patients increased as the risk of the abscissa increased,and that the survival life of the deceased patients,represented by the red circle chart,was more brief than that of the surviving patients,represented by the blue circle chart.In the test group,low risk patients tended to have higher OS than high risk patients,but the distinction was not statistically significant (P=0.089;Figure 2K),possibly caused by the small sample size.Following that,the survival analysis of the three groups revealed that the low risk patients exceeded the OS in the training and all sets (P< 0.001;Figure 2J and L).The aforementioned findings demonstrate how well the risk score model predicts patient survival.The concentrations of AL354993.2,AC006213.7,AC013652.1,and AL683813.1 were more abundant in the high-risk group,according to an analysis of the risk score heatmap of the two groups.This suggested that these four DRLs may act as biomarkers for poor CRC prognosis.Comparing the high-risk group to the low-risk group revealed that AC093157.1,AC005034.5,TNFRSF10A-AS1,and AC011815.1 were less abundant,suggesting that these four DRLs may be valuable prognostic indicators for CRC (Figure 2G-I).All patients were also subjected to a progression-free survival analysis,with the results demonstrating that the low-risk group had a longer period of high-quality survival (Figure 2M).This observation is consistent with the finding that the low-risk group had a higher OS than the high-risk group.In addition,based on clinical factors such age,gender,and stage,we then compared the chance of survival and clinical traits of CRC patients.These findings demonstrated that high-risk patients had shorter OS than low-risk patients regardless of clinical characteristics,with the exception of stage I-II (Figure 3).We also note that inconsistency in results may be due to the small patient population within this group and the poor prognosis associated with advanced CRC,both of which may impact the accuracy of results for stage I-stage II patients.In conclusion,the clinical outcomes of CRC patients can be predicted using a prognostic model developed using on DRLs risk assessment.Patients’ DRLs risk scores were negatively associated with OS,with greater risk scores being accompanied by shorter OS and a poorer outlook.
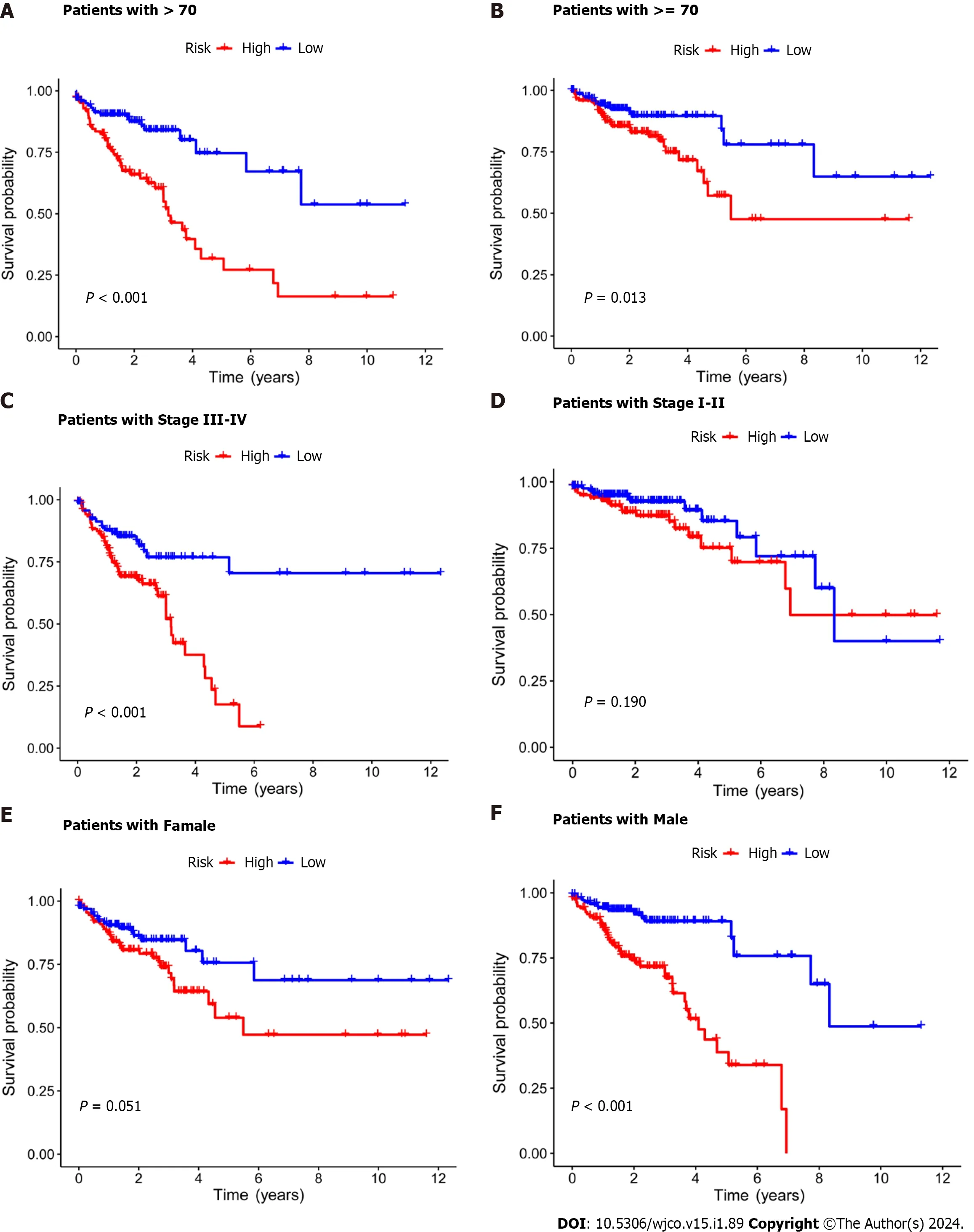
Figure 3 Kaplan-Meier survival analysis of high-and low-risk patients based on different clinical variables. A and B: Age;C and D: Stage;E and F: Gender.
DRLs risk prognosis model is a robust determinant of clinical outcomes of CRC patients
Patient age,gender,cancer stage,and risk score were subjected to single-and multi-factor Cox regression analyses.We discovered that both methods indicated risk scores that were statistically significant (P< 0.001),indicating that risk score is an independent prognostic factor for CRC apart from other clinical variables (Figure 4A and B).The areas under the curve (AUCs) for the 1-,3-,and 5-year ROCs curves were also plotted (Figure 4C) indicates the high accuracy of the DRLs risk prognostic model for predicting the OS of patients.Furthermore,the AUC of the risk score was 0.665 (Figure 4D),which shows that the model’s predictive power is greater than the additional clinical variables besides staging.Consistent with these findings,the C-index curve (Figure 4E) also revealed that the risk model had a higher concordance index than all clinical factors other than staging.A calibration plot was then created to compute OSviarisk score and patient clinical parameters.According to these findings,patients had survival rates of 0.933,0.803,and 0.722 after one,three,and five years,respectively (Figure 4F).A calibration curve (Figure 4G) verified the accuracy of the calibration plot.The DRLs risk prognostic model,in conclusion,reliably predicts the CRC patient survival and functions as an exceptional predictive indicator that is independent of other clinical characteristics.
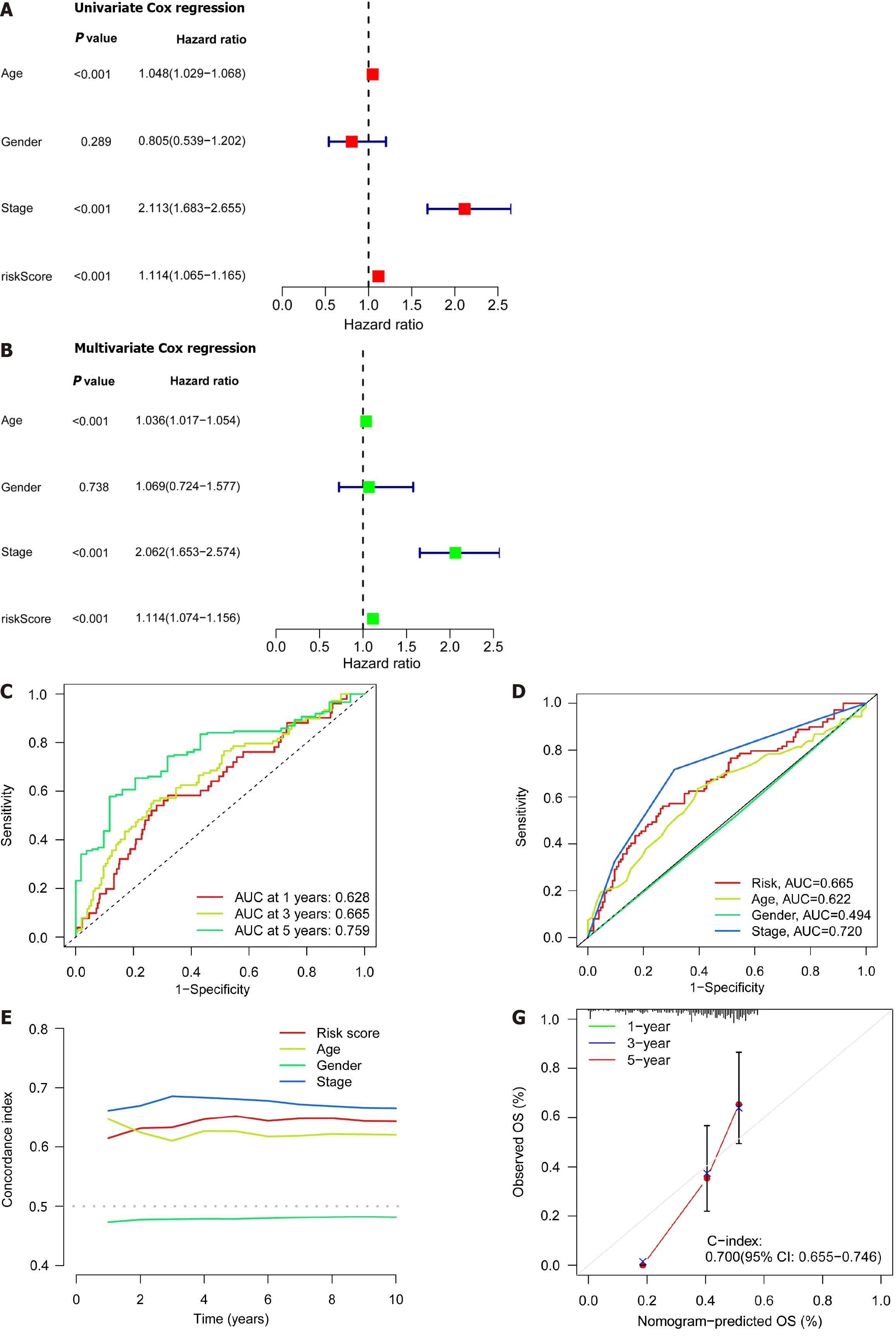
Figure 4 lndependent prognostic ability and predictability of clinical outcomes of the disulfidaptosis-related lncRNA risk scoring model. A: Univariate Cox regression analysis of clinical variables and risk scores;B: Multivariate Cox regression analysis of clinical variables and risk scores;C: Prediction of 1-,3-,and 5-year overall survival (OS) for all enrolled colorectal cancer (CRC) patients;D: Comparison of risk scoring model and clinical variables in predicting OS in CRC patients;E: C-index ROC curve of the risk model;F: Column chart of risk and clinical variable features predicting 1,3,and 5-year OS in CRC patients;G: Calibration curves demonstrate the accuracy of the risk model in predicting 1,3,and 5-year OS in CRC patients.
GO, KEGG, and GSEA
DEGs were identified using the average gene expression levels of samples from the high-and low-risk group samples (Padj < 0.05,|log2 (fold change)|1,Supplementary Table 4).We used the “Bioconductor” R package in R software to perform GO enrichment analysis and KEGG pathway analysis to investigate the biological roles of DEGs.The biological processes (BP) that DEGs were found to be involved in included “chromatin remodeling,” “protein-DNA complex subunit organization,” “nucleosome organization,” and “positive regulation of secretion,” among others.We also observed significant increases in the expression of DEGs annotated as “DNA packaging complex,” “protein-DNA complex,” “nucleosome,” and “endoplasmic reticulum lumen” have been noted in cellular components (CC).DEGs have been correlated to "signaling receptor activator activity,” “receptor ligand activity,” “peptidase regulator activity,” and “peptidase inhibitor activity,” among other molecular functions (MF;Figure 5A and 5B,Supplementary Table 5).These findings suggest that DEGs significantly participate in the control of the immunological response.In addition,KEGG pathway analysis showed that DEGs were primarily involved in “Neutrophil extracellular trap formation,” “IL-17 signaling pathway,” and “PPAR signaling pathway” (Figure 5C).The biological pathways “skeletal system development” and “Cell surface receptor signaling pathway involved in cell-cell signaling” were also discovered to be activated in the high-risk group by GSEA enrichment analysis.In terms of CC,enrichment was observed in the “Collagen-containing extracellular matrix” and "Endoplasmic reticulum lumen” terms.The biological functions “Nucleosome assembly” and “Nucleosome organization" were higher in the low-risk population,as well as the biological elements “DNA packaging complex” and “Nucleosome” (Figure 5D and E).
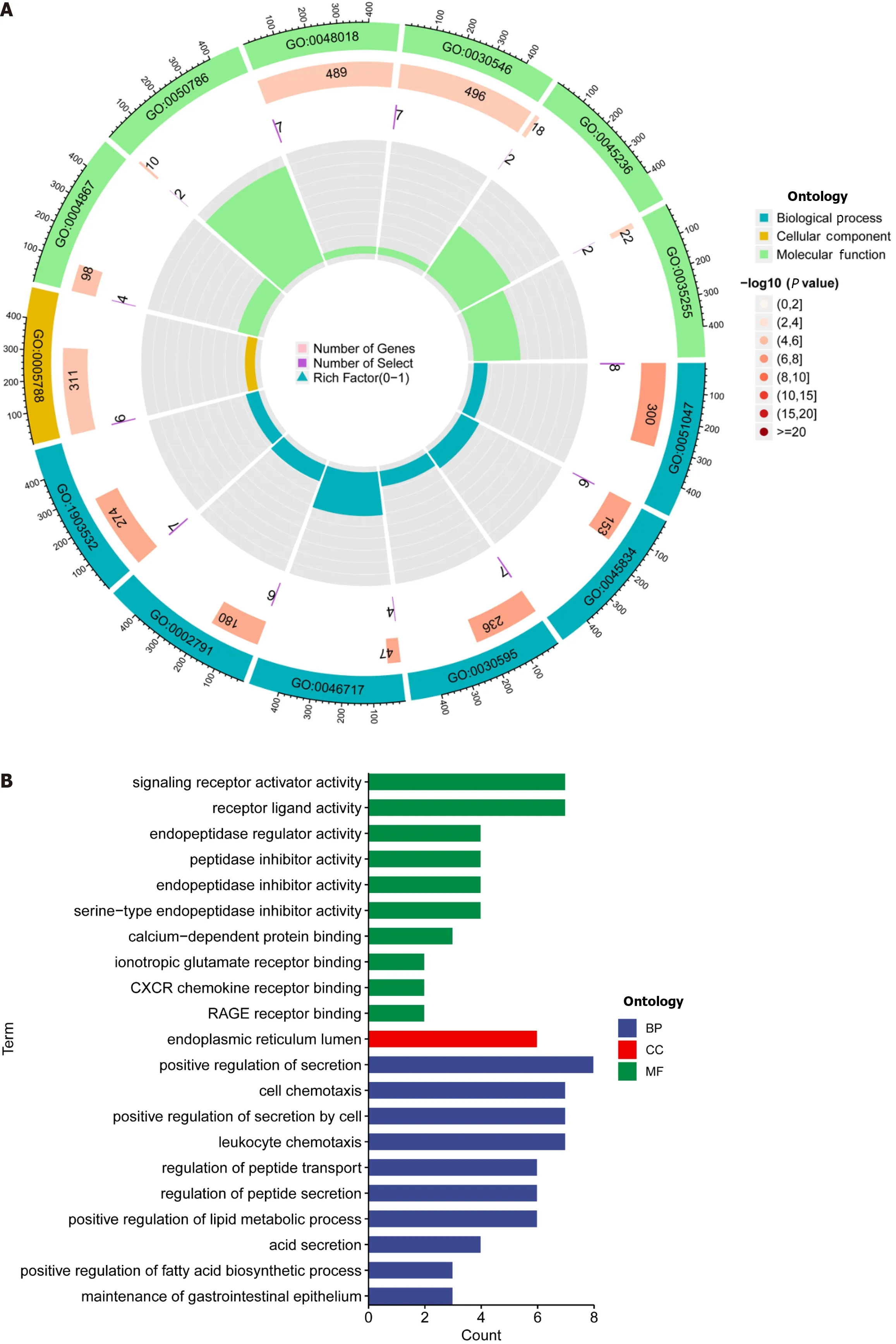
Figure 5 Enrichment analysis using the gene ontology,Kyoto encyclopedia of genes and genomes,and gene sets using gene set enrichment analysis frameworks. A and B: Gene ontology analysis reveals the diversity of molecular biology processes,cellular components,and molecular functions;C: Kyoto encyclopedia of genes and genomes pathway analysis identifies significantly enriched pathways;D and E: The top five pathways enriched between high and low-risk populations as identified by gene set enrichment analysisanalysis.
Analysis of the tumor immune microenvironment for high- and low-risk CRC patients
The tumor immune microenvironment controls immune-related tumors in CRC.Immunological cell subsets in CRC have many roles,and may exert either immunosuppressive or antitumor immunological effects to accelerate tumor growth[31].The percentages of immune cells that infiltrate tumors were then calculated for the high-and low-risk groups (Supplementary Table 6).We discovered that the high-risk group had a lower percentage of inactive T cells with CD4 memory,inactive dendritic cells,active dendritic cells,and active eosinophils relative to the low-risk group.Moreover,compared to the high-risk group,the low-risk group had lower percentages of regulatory T cells (Tregs),dormant NK cells,and M0 macrophages (Figure 6A).Gene Set Variation Analysis (GSVA) is a gene set enrichment method that evaluates differences between different samples by performing pathway-centric analysis of gene sets.Next,using GSVA,we then examined variation in immune-related activities between high-and low-risk groups.According to these findings,the low-risk group was more substantially connected with cytolytic activity,MHC class I,and neutrophils (Figure 6B).Moreover,a correlational study between the quantity of tumor-infiltrating immune cells and the eight important DRLs revealed a positive link between plasma cells,CD8 T cells,regulatory T cells (Tregs),and the eight critical DRLs (Figure 6C).In addition,we conducted a correlational analysis between immunological checkpoints and risk scores.This revealed a positive association between cancer associated fibroblast (CAF) and M2-like tumor-associated macrophage (TAM M2),a negative correlation between interferon gamma (IFNG) and CD274,and no significant correlation between these correlations (Figure 6D).Taken together,these findings imply that the tumor immunological milieu in the high-and low-risk groups differs significantly.Due to its strong immune surveillance position,the low-risk group displays a higher abundance of resting immune cells.Moreover,T cells,NK cells,and other immune cells,which are linked to tumor invasion,metastasis,and CRC development,are present in higher amounts in the high-risk group.
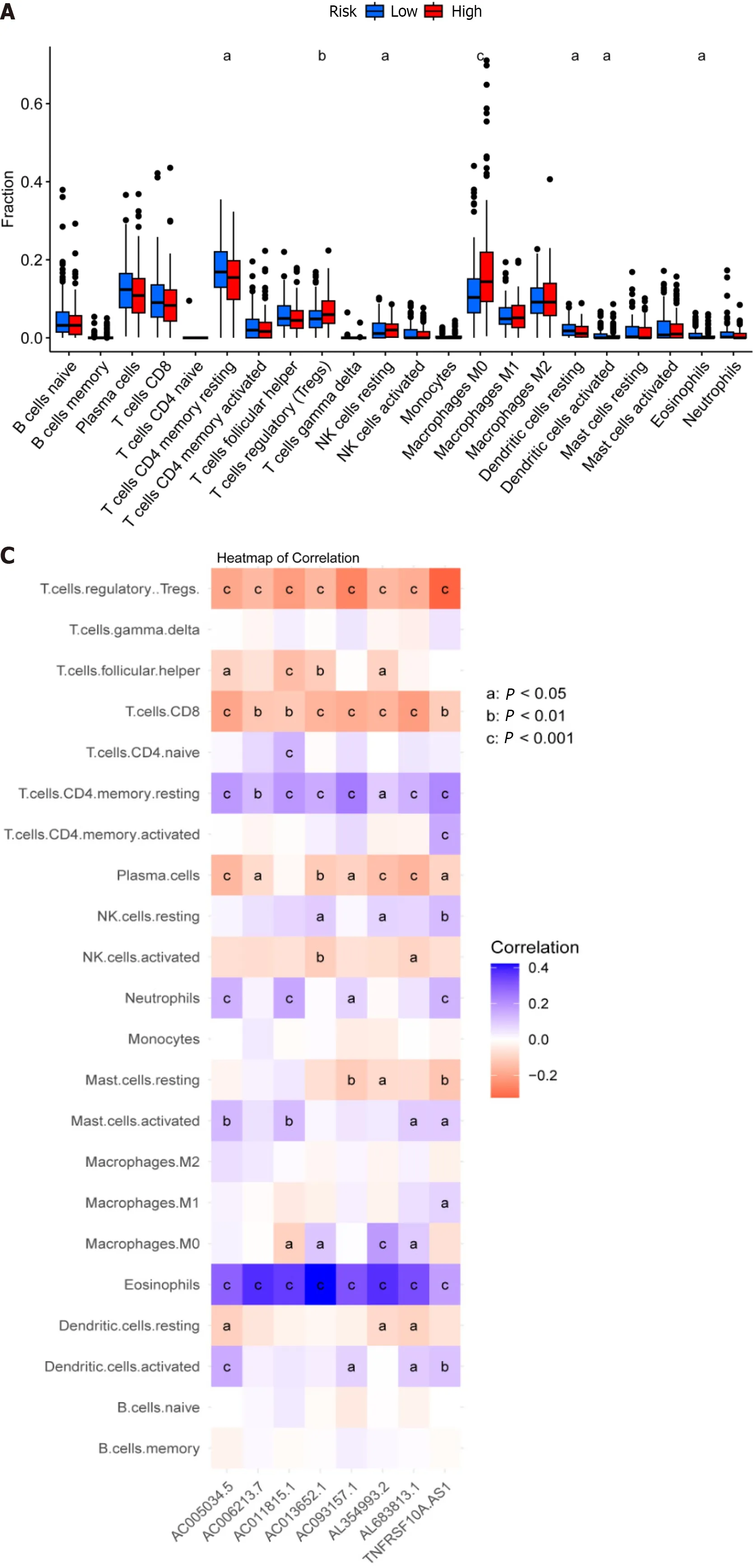
Figure 6 Analysis of the tumor immune microenvironment for high-and low-risk patient groups. A: Shown are (A) Violin plots of the proportions of 22 tumor-infiltrating immune cells;B: Differential analysis of immune-related functions between high-and low-risk groups;C: Correlation analysis between infiltrating immune cells and eight key disulfidaptosis-related lncRNAs (disease risk factors);D: Correlation analysis between immune checkpoints and risk scores.aP < 0.05;bP < 0.01;cP < 0.001.
CRC mutation landscape, differential analysis of TMB, and TIDE
Next,we retrieved somatic mutation data from the TCGA database and generated waterfall plots to conduct intergroup comparisons using the maftools R package.This analysis was performed to investigate genetic alterations in patients from the high-and low-risk CRC groups.These plots revealed the existence of 15 highly altered genes,including APC,TP53,TTN,KRAS,MUC16,SYNE1,PIK3CA,FAT4,RYR2,ZFHX4,OBSCN,DNAH5,LRP1B,and CSMD3.APC,TP53,and TTN were found to have higher mutation frequencies in the high-risk population compared to the low-risk population,whereas MUC16,SYNE1,LRP1B,CSMD3,and CSMD1 showed higher mutation rates in the low-risk population (Figure 7A and B).The immunotherapy response has been linked to TMB[32],and here the low-risk group showed a larger mutation burden according to differential analysis of TMB (Figure 7C).The term TIDE describes a tumor cell’s capacity to elude immune monitoring and suppress an immunological response;this can happenviaa variety of methods.In contrast to the low-risk group,the high-risk group showed a higher TIDE score,as shown in Figure 7D.These findings demonstrate that immune evasion and tumor cell mutations are more common in the low-risk group of CRC patients,suggesting that there is a substantial therapeutic potential for immune checkpoint inhibitors for the management of lowrisk CRC.
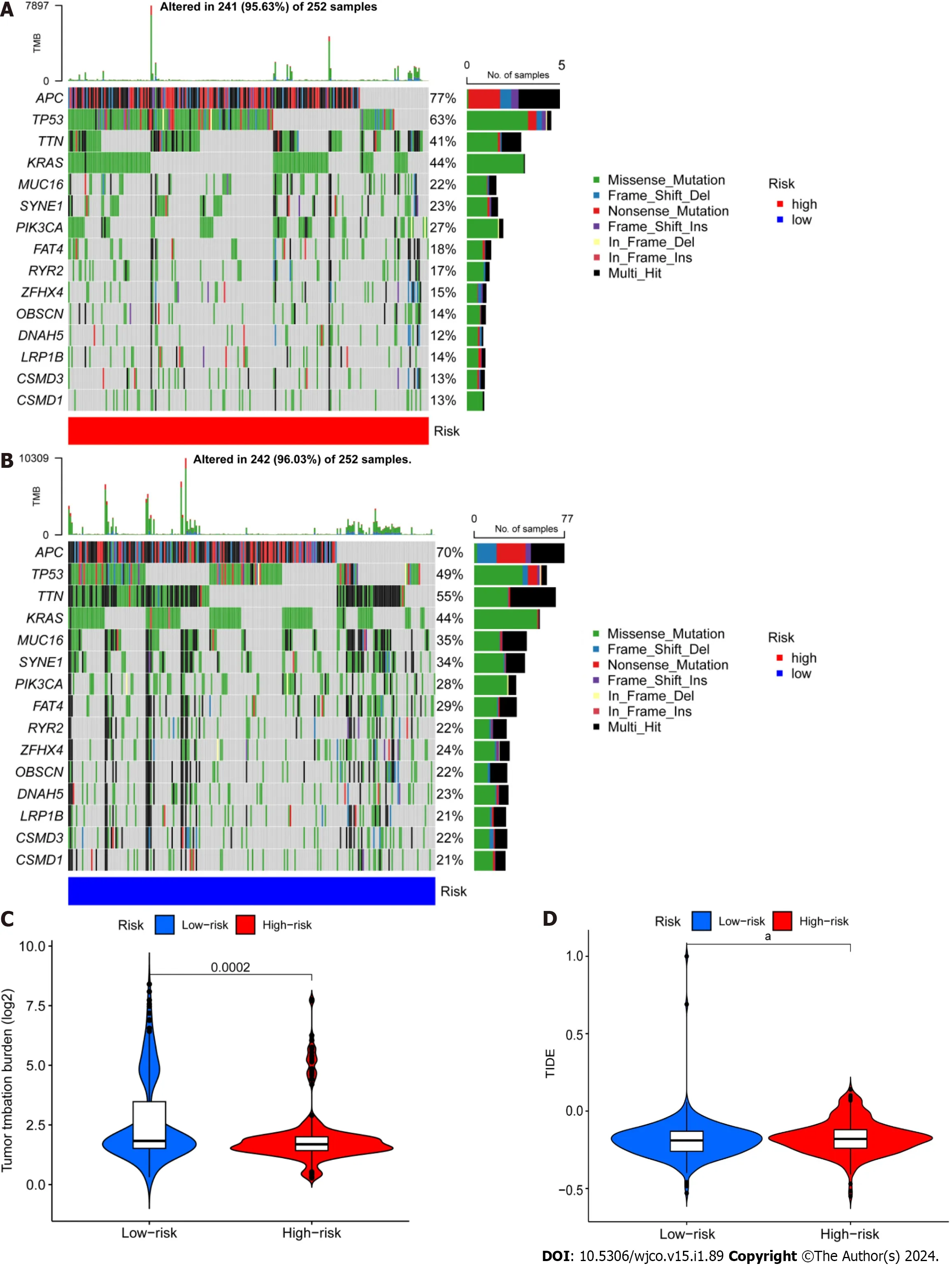
Figure 7 Differential analysis of tumor mutation burden and tumor immune dysfunction and exclusion. A and B: Waterfall plots depicting 15 highly mutated genes in the high-and low-risk colorectal cancer (CRC) groups;C: Differential analysis of TMB in patients from the high-and low-risk CRC groups;D: Tumor immune dysfunction and exclusion analysis in patients from the high and low-risk groups.aP < 0.05;bP < 0.01;cP < 0.001.
Identification of potential drugs for CRC
GDSC project is a database that focuses on the molecular indicators of therapeutic response and drug sensitivity in cancer cells.It is used to find novel therapeutic cancer biomarkers[33].Here,an analysis using the oncoPredict identified four medicinesi.e.,epirubicin,bortezomib,teniposide,and BMS-754807 that showed limited sensitivity toward CRC (Figure 8A-D).Of these,Forkhead box protein p3 (Foxp3) modulates epirubicin and has been identified as a suppressor of Treg cell function[34].Moreover,the proteasome inhibitor bortezomib prevents CRC cells from forming spheres and thereby from self-renewing by inhibiting the fundamental transcription factor CTNNB1[35].Teniposide,a topoisomerase II inhibitor,also strongly affects CRC gene expression[36].Finally,the insulin-like growth factor 1 receptor (IGF-1R) inhibitor BMS-754807 has been found to have an impact on the growth and survival of tumor cells[37].Drug sensitivity (also known as the half-maximal inhibitory concentration,or IC50is the degree to which an organism responds to a particular drug,and the potency of the medicine decreases as the IC50value rises.Epirubicin,Bortezomib,and teniposide all demonstrated greater sensitivity in the high-risk group,which suggests that the low-risk group may benefit from greater efficacy.The high-risk group,however,benefited the most from BMS-754807.
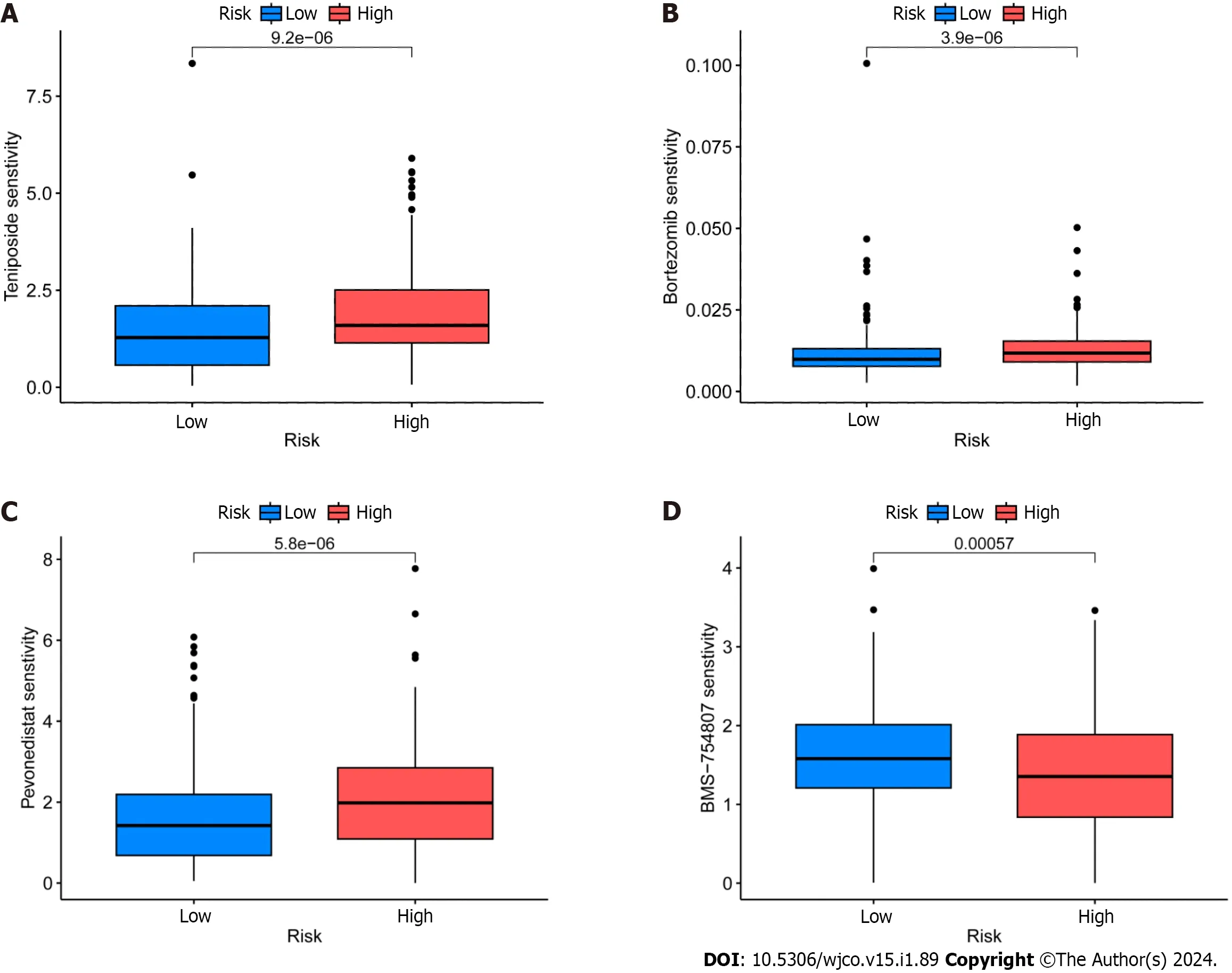
Figure 8 ldentification of potential drugs for the treatment of colorectal cancer. A: Epirubicin;B: Borte zomib;C: TeniposideD: BMS-754807.
Validation of disulfidoptosis-related features in NCM460 and HT-29 cells
In this study,through multifactorial Cox regression analysis,we identified eight Disulfidoptosis-related lncRNAs that exhibited differential expression in CRC prognosis within the TCGA-COAD and TCGA-READ datasets.These lncRNAs include AL354993.2,AC006213.7,AC013652.1,AL683813.1,AC093157.1,AC005034.5,TNFRSF10A-AS1,and AC011815.1.Among these,the first four showed higher expression in the high-risk group associated with poor prognosis,while the latter four displayed an inverse trend.To validate the expression of these eight prognosis-related lncRNAs,we conducted qPCR experiments in two cell lines: normal colonic epithelial cells NCM460 and colorectal tumor cells HT-29.Our experiments revealed that AL354993.2,AL683813.1,AC093157.1,AC005034.5,and AC011815.1 exhibited significant differences in expression levels between HT-29 and NCM460 cells,with the former displaying higher expression levels.AC013652.1 and TNFRSF10A-AS1 exhibited a trending pattern but did not demonstrate significant differences (Figure 9).AC006213.7 was not found in the lncRNA database,and therefore,we do not discuss its expression levels at this time.TCAP,NNAT,CHGB,and COL2A1 were among the top four differentially expressed genes between the high and lowrisk groups,with higher abundance in the high-risk group.Using qPCR technology,we also assessed the mRNA levels of these four genes and found that their expression was higher in HT-29 cells compared to NCM460 cells.
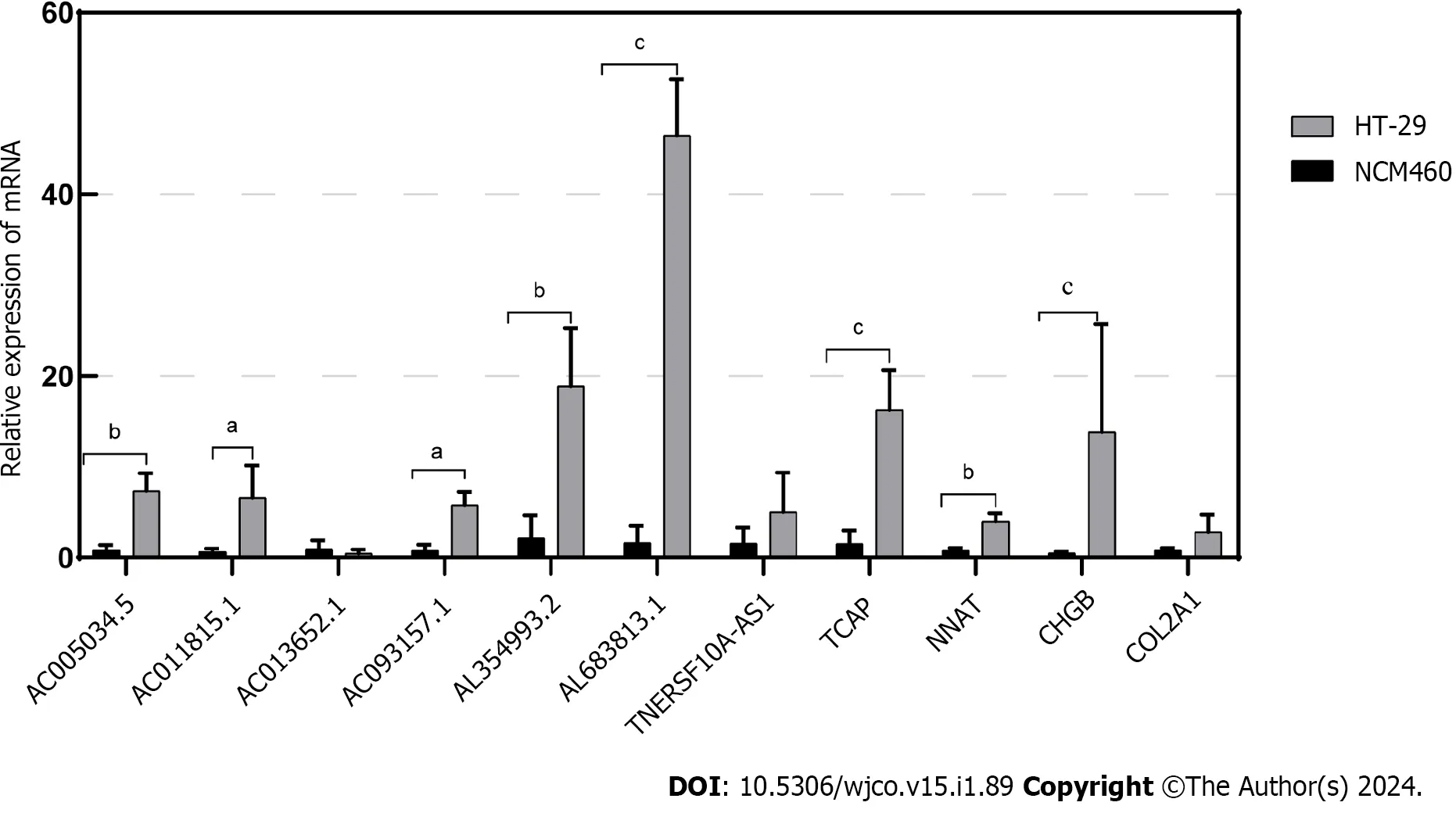
Figure 9 Validation of the relationship between lncRNAs/Genes and cell death induced by disulfidoptosis. The expression of lncRNAs/Genes in NCM460 and HT-29 cells was measured by quantitative real-time polymerase chain reaction (n=3).aP < 0.05;bP < 0.01;cP < 0.001.
Induction of the disulfidoptosis cell model and in vitro validation of the risk prediction model
We constructed a disulfidoptosis cell model to investigate changes in the mRNA expression levels of LncRNAs in the CRC risk prediction model.Different concentrations of WZB117 (0,1,3,10,15,30,50,100,300 µmol/L) were applied to HT-29 cells to assess cell viability.As shown in Figure 10,cell viability was significantly inhibited at WZB117 concentrations of 50 µmol,100 µmol,and 300 µmol.Therefore,we selected 300 µmol as the effective induction concentration for Disulfidoptosis.Q-PCR results indicated that after WZB117 induction at 300 µmol,AL354993.2,AC013652.1,and AL683813.1 were upregulated in HT-29 cells,while AC005034.5,TNFRSF10A-AS1,and AC011815.1 were downregulated.The expression of TCAP,NNAT,CHGB,and COL2A1 was upregulated (Figure 11).Surprisingly,AC093157.1 increased after WZB117 induction,which was contrary to the risk model trend,possibly due to cell line-specific factors.

Figure 10 Cell viability assessment of HT-29 cells treated with WZB117 at different concentrations for 24 h.
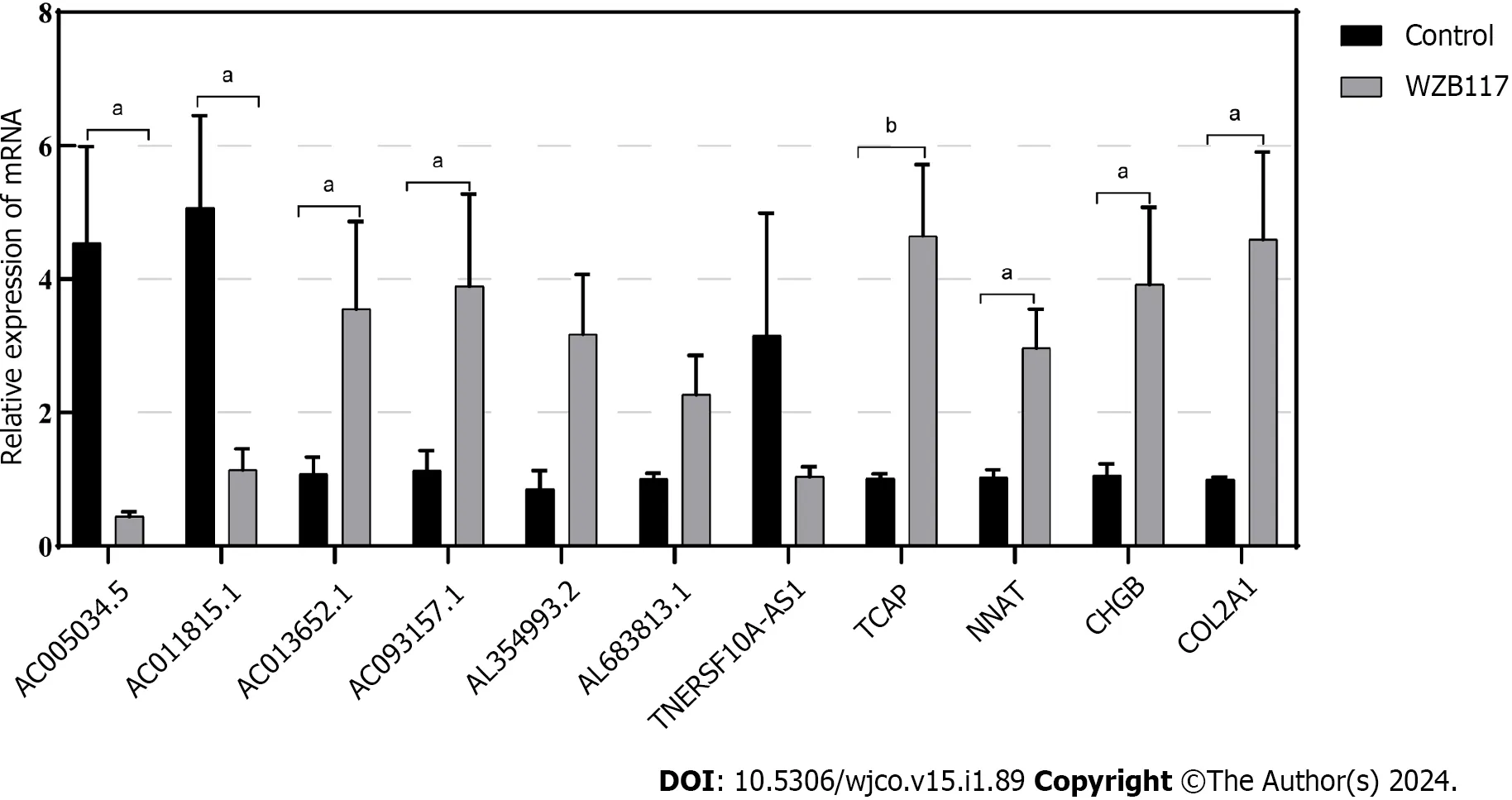
Figure 11 Validation of the relationship between lncRNAs/Genes and cell death induced by disulfidoptosis. The expression of lncRNAs/Genes in HT-29 cells was measured by quantitative real-time polymerase chain reaction (n=3) after treatment with 300 µmol/L WZB117 for 24 h.aP < 0.05;bP < 0.01;cP < 0.001.
In summary,Disulfidoptosis can influence the expression of LncRNAs and differential genes in the risk prediction model.Therefore,these key LncRNAs and differential genes have the potential to serve as diagnostic markers for CRC,aiding in the treatment and prediction of the degree of CRC disease risk.
DlSCUSSlON
CRC tumors have unique characteristics,including a high incidence,a high rate of metastasis,and a high fatality rate.Moreover,both the aberrant expression and regulation of multiple genes are involved in its onset and development.lncRNAs,which participate in vital BP such cell proliferation,apoptosis,invasion,and metastasis,have been demonstrated to play significant roles in CRC.For instance,the overexpression of LncRNA LINC00460 triggers the epithelial-mesenchymal transition and aids in the development of cancer.In CRC,LINC00460 interacts with the ATPdependent RNA helicase A (DHX9) and insulin-like growth factor 2 mRNA-binding protein 2 (IGF2BP2) to recognize the high mobility group AT-hook 1 (HMGA1) and control its m6A modification.This in turn controls HMGA1 expression,improves mRNA stability,and aids tumor metastasis[38].This suggests that lncRNAs may be useful therapeutic targets for CRC.Moreover,a novel type of cell death called disulfidptosis is known to take place under conditions of glucose starvation.Related to this process,SLC7A11 is upregulated in CRC,which speeds up the loss of cytoplasmic NADPH,thereby causing disulfide linkages to form in protein molecules and instigating the collapse of the actin network and cytoskeleton,finally resulting in cell death[39].This study therefore analyzes the roles and regulatory mechanisms of lncRNAs connected to disulfidptosis in CRC,and offers new targets and methods for determining CRC prognosis.
Through bioinformatics analyses,we discovered eight key DRLs: AC005034.5,AC006213.7,AC011815.1,AC013652.1,AC093157.1,AL354993.2,AL683813.1,and TNFRSF10A-AS1.Previous reports suggested that the osteosarcoma protective factor AC005034.5 is downregulated in cases of increased disease risk[40].Moreover,the elevated abundance of AC013652.1,a key prognostic marker for colorectal and stomach malignancy,predicts a poor prognosis for the patient[41,42].By facilitating cell apoptosis and controlling immune cell infiltrationviathe overexpression of zinc finger protein 268[43],AC093157.1 has been shown to slow the development of clear cell renal cell carcinoma.In addition,the ability of gastric cancer cells to proliferate,advance through the cell cycle,and invade other tissues has been found to be strongly enhanced by TNFRSF10A-AS1[44].Here,we created a risk prognosis model that divides patients into high-and low-risk groups based on these eight key DRLs.Using column plots,ROC curve analysis,c-indexes,calibration curves,and univariate and multivariate Cox regression analyses,we then validated the robustness and independence of the model (Figure 4).Next,we used the model’s risk scores to contrast the survival rates of high-and low-risk groups.We observed that increased risk scores were associated with higher mortality in CRC patients (Figure 2D-F).This result shows that the risk score can be used as a reliable measure to predict patient survival.To enhance the reliability of the risk prediction model,we assessed the mRNA expression levels of the 8 Disulfidoptosis-Related LncRNAs (DRLs) in two cell lines,NCM460 and HT-29.Subsequently,induction of the Disulfidoptosis cell model using the Glut1 inhibitor WZB117 demonstrated that the accumulation of disulfides leads to increased cell death and alters the expression levels of DRLs.
Next,GO enrichment analysis of DEGs from the high-and low-risk groups revealed that DEGs were primarily engaged in the immune response.DRLs were strongly enriched in KEGG annotation terms for “Neutrophil extracellular trap formation (NET),” “IL-17 signaling pathway,” and “PPAR signaling pathway.” According to GSEA,the BP “Cell surface receptor signaling pathway involved in cell-cell signaling” and “skeletal system development” were active in the high-risk group (Figure 5).Releasing chromatin DNA threads encircling granule proteins,neutrophils release NET to capture microorganisms.Research points to a connection between NET growth and cancer pathogenesis.Vascular NETs can increase vascular permeability,which makes it easier for cancer cells to enter organs from vessels.Via its transmembrane receptor,the transmembrane protein CCDC25,NET-DNA performs as a chemoattractant for cancer cells.Through activating the ILK-β-parvin pathway,this interaction improves the motility of tumor cells[45].The IL-17 signaling pathway is hazardous for the occurrence of cancer and strongly related to the advancement of inflammation.Adenomatous polyposis coli (Apc)-carrying intestinal epithelial cells proliferate in response to IL-17 signaling in the intestine,which facilitates the production of adenomas[46].Intestinal barrier function is compromised by adenomas,which additionally increase IL-17 responses in tumors and hence accelerate tumor growth[47].The ligand-activated nuclear receptor PPAR influences energy homeostasis and lipid metabolism[48].Findings indicates that CRC tumor cells have abnormal activation of the PPAR signaling system.When PPARγ inhibitors are administered for blocking this pathway,tumor epithelial cell proliferation is dramatically suppressed and apoptosis is increased[49].Here,the low-risk group exhibited a larger mutational load and a lower TIDE score during the study of differences in TMB (Figure 7).This suggests that the low-risk group may respond better to immune checkpoint inhibitor medication and has a lower chance of immunological escape.Furthermore,relative to the high-risk group,the expression levels of the immunological checkpoints CD274 (PD-L1) and IFNG were higher in the low-risk group.Conversely,the high-risk group exhibited higher levels of CAF and TAM M2 expression.However,additional experimental validation is necessary to ascertain whether these checkpoint inhibitors can be used as antitumor medications for CRC.Previous research has shown that solid CRC tumors typically exhibit disruptions in the IFNG-JAK-STAT-TET signaling pathway,which facilitates anti-PDL1/PD-1 immunotherapy.Furthermore,IFNG is an important antiangiogenic mediator of tumor immunity.As a result,CD274 and IFNG may serve as promising targets for the therapy of CRC[50].Finally,we showed that epirubicin,Bortezomib,teniposide,and BMS-754807 represent four possible CRC therapy medicines.Drug sensitivity results revealed that the first three medications showed greater efficacy in the low-risk group,while BMS-754807 was better suited to treat the high-risk group (Figure 8).Although teniposide has not yet undergone clinical trials for colorectal cancer,a check of the ClinicalTrials.gov database (https://clinicaltrials.gov) indicated that epirubicin,Bortezomib,and BMS-754807 are currently involved in several CRC-related clinical trials.We anticipate clinical study results for these four medications in the hope that they will show beneficial therapeutic benefits for the treatment of CRC.
As a result,the findings of the research we conducted highlight the complex role that disulfidptosis plays in the onset and course of colorectal cancer.We created a prognostic risk assessment feature by utilizing DRLs.This model illustrates the processes behind cellular disulfidptosis and predicts CRC patients' prognosis with consistency.Relevant pathways correlated to CRC immunity and prognosis were found by our analysis of genes that are DEGs in high-and low-risk sample groups." This the discovery holds significant therapeutic implications for both the prevention and the treatment of CRC,as it is the first in the history of CRC research to combine disulfidptosis,LncRNA,and immunotherapy.However,there are limitations related to our database selection and analysis.Firstly,the sample size was small in our study.A study using a bigger sample size should be conducted in the future.Secondly,all data sources were derived from the TCGA database,which lacks significant experimental and clinical data to evaluate the accuracy of our results.Finally to confirm the functional properties of DEGs and the anticancer mechanisms of immunological checkpoints,we must refine the experimental design in future studies.
CONCLUSlON
In conclusion,this is the first attempt to apply bioinformatics methods to examine the immune cell infiltration and the expression patterns of LncRNAs associated with the disulfidptosis genes in high-and low-risk groups of colorectal cancer patients.It is stressed the significance of LncRNAs in the diagnosis and treatment of colorectal cancer.Disulfidptosis is the name given to the accumulation of intracellular disulfide compounds that leads to the breakdown of cytoskeletal proteins,which may affect host homeostasis and promote tumor growth.The eight significant LncRNAs that have been discovered are closely associated to the disulfidptosis genes,which can be used as prognostic indicators to predict the clinical course of colorectal cancer patient.Furthermore,they have the capacity to act as regulators to restrain the development,differentiation,invasion,and metastasis of cancer cells,providing fresh approaches for the focused treatment of colorectal cancer.
ARTlCLE HlGHLlGHTS
Research background
Colorectal cancer (CRC) is an extremely fatal disease that is the third fastest-growing cause of cancer-related death globally.Disulfidptosis is one particular type of cell death that has been associated to the growth,escape,and regeneration of cancer cells.With disulfidptosis,colorectal cancer treatments and survival predictions could be altered.
Research motivation
A large number of clinical studies incorporate statistical significance to present their results.However,to be able to assess a therapy's adaptability and relevance in routine clinical practice,clinical measurements of significance are necessary.
Research objectives
The main goal of this work is to construct a stable biological biomarker that utilizes long non-coding RNA (LncRNA) linked to disulfidptosis-induced cell death.This may provide innovative viewpoints on the assessment of immunotherapy response and prognosis in patients suffering from CRC.
Research methods
The Cancer Genome Atlas (TCGA) database offered transcriptome,clinical,and genetic mutation data relating to CRC.The minimal absolute shrinkage and selection operator approach and univariate and multivariate Cox regression models were applied to discover and assess critical LncRNA correlated with disulfidptosis.Ultimately,the critical LncRNA served as the foundation for the prognostic model.
Research results
Through multivariate analysis,we succeeded to identify eight critical long non-coding RNAs linked to disulfidptosis.These LncRNAs had significant accuracy for the consequences of CRCs.Compared to the high-risk group,patients in the low-risk group had a higher rate of overall survival.As a result,the nomogram prediction model we created exhibits good predictive validity and incorporates clinical characteristics and risk scores.
Research conclusions
As a way to predict the prognosis of patients with colorectal cancer,we constructed a prediction model of disulfidptosisrelated LncRNAs based on the TCGA-COAD and TCGA-READ cohort using bioinformatics technology and clinical patient data.The application of this model in clinical practice makes it much simpler to classify CRC patients precisely,pinpoint subgroups that are more likely to benefit from immunotherapy and radiation therapy,and provide evidencebased,targeted therapies for CRC patients.
Research perspectives
In subsequent research,we must enhance the animal and cell experiments in order to validate the functional characteristics of disulfidaptosis-related lncRNA and the immune checkpoints' anticancer mechanisms.
ACKNOWLEDGEMENTS
We are very grateful for data provided by databases such as TCGA.Thanks to reviewers and editors for their sincere comments.
FOOTNOTES
Author contributions:Chen YG provided the acquisition of funding and formulated research goals;Wang KL wrote the original manuscript;Chen KD,and Chen ZP were involved in the software analysis;Tang WW,Wang YJ,and Shi GP performed the data collation;All authors have read and agreed to the published version of the manuscript.
Supported byJiangsu Province Science and Technology Plan Project-Youth Fund Project,No.BK2020040973.
lnstitutional review board statement:The ethical approval is not applicable to this article.
lnformed consent statement:There were no human subjects included in this article,and therefore informed consent is not applicable.
Conflict-of-interest statement:All the authors declare that the study was carried out without any commercial or financial relationships which could be considered a potential conflict of interest.
Data sharing statement:The original contributions presented in the study are included in the article.Further inquiries can be directed to the corresponding authors.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers.It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BY-NC 4.0) license,which permits others to distribute,remix,adapt,build upon this work non-commercially,and license their derivative works on different terms,provided the original work is properly cited and the use is non-commercial.See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:China
ORClD number:Yu-Gen Chen 0000-0001-9221-218X.
S-Editor:Liu JH
L-Editor:A
P-Editor:Zhang XD
杂志排行

World Journal of Clinical Oncology的其它文章
- Prognostic factors of breast cancer brain metastasis
- Radiotherapy for hyoid bone metastasis from lung adenocarcinoma: A case report
- What are the changes in the hotspots and frontiers of microRNAs in hepatocellular carcinoma over the past decade?
- Fatty acid binding protein 5 is a novel therapeutic target for hepatocellular carcinoma
- Gene signatures to therapeutics: Assessing the potential of ivermectin against t(4;14) multiple myeloma
- ldentification of the key genes and mechanisms associated with transcatheter arterial chemoembolisation refractoriness in hepatocellular carcinoma