胃肠道透明细胞肉瘤样肿瘤/恶性胃肠道神经外胚层肿瘤2例临床病理学特征
2024-03-07李解珍黄海建陈灵锋吴义娟陈小岩
李解珍,黄海建,曾 强,刘 霖,陈灵锋,吴义娟,陈小岩
胃肠道透明细胞肉瘤样肿瘤/恶性胃肠道神经外胚层肿瘤(clear cell sarcoma-like tumor of the gastrointestinal tract, CCSLTGT/ malignant gastrointestinal neuroectodermal tumor, MGNET)属于非常罕见的间叶源性肿瘤[1-2],全球仅报道120例。CCSLTGT/MGNET具有上皮样和(或)梭形细胞形态,易被误诊为其他常见原发性胃肠道肿瘤,术前诊断困难。本文收集2例CCSLTGT/ MGNET的临床资料,分析其临床病理学和分子学特征,为临床诊断、治疗和预后提供依据。
1 材料与方法
1.1 临床资料收集2例胃肠道CCSLTGT/MGNET(表1),患者均为男性,年龄分别为22、47岁,临床表现为腹痛(2/2),腹部肿块(2/2),伴排便次数增多(2/2);伴血便(2/2),发热(1/2)。影像学检查:受累肠壁见实性软组织肿块阴影,增强后呈不均匀强化。

表1 2例胃肠道透明细胞肉瘤样肿瘤/恶性胃肠道神经外胚层肿瘤临床病理特点
1.2 方法组织标本均经10%中性福尔马林固定,常规制片,行HE和免疫组化EnVision法染色,一抗SOX10、S-100、Syn、CgA、CD56、HMB-45、Melan A、Fli-1、NKX2.2、CD99、H3K27Me3、INI1、desmin、MyoD1、Myogenin、CKpan、CD34、CD117、DOG1、PD-L1和Ki67,均购自福州迈新公司。2例均采用FISH法检测EWSR1基因分离,试剂盒购自美国雅培Abbott公司,具体操作步骤严格按试剂盒说明书进行。1例同时行二代测序(next generation sequencing, NGS)基因检测,采用目标区域探针捕获技术和基于Illumina测序平台的NGS对样本进行检测。
2 结果
2.1 眼观2例均为隆起型肿物,切面灰白色,质地中等。
2.2 镜检2例形态学相似,低倍镜下见肿瘤细胞呈实性、片状及巢状排列(图1),部分区域可见菊形团样结构(图2);高倍镜下见瘤细胞呈卵圆形、短梭形或上皮样,胞质嗜酸性、局灶透明,未见胞质内黑色素(图3)。核圆形,部分可见小核仁,核分裂象8~10个/10 HPF,可见散在分布破骨样多核巨细胞。
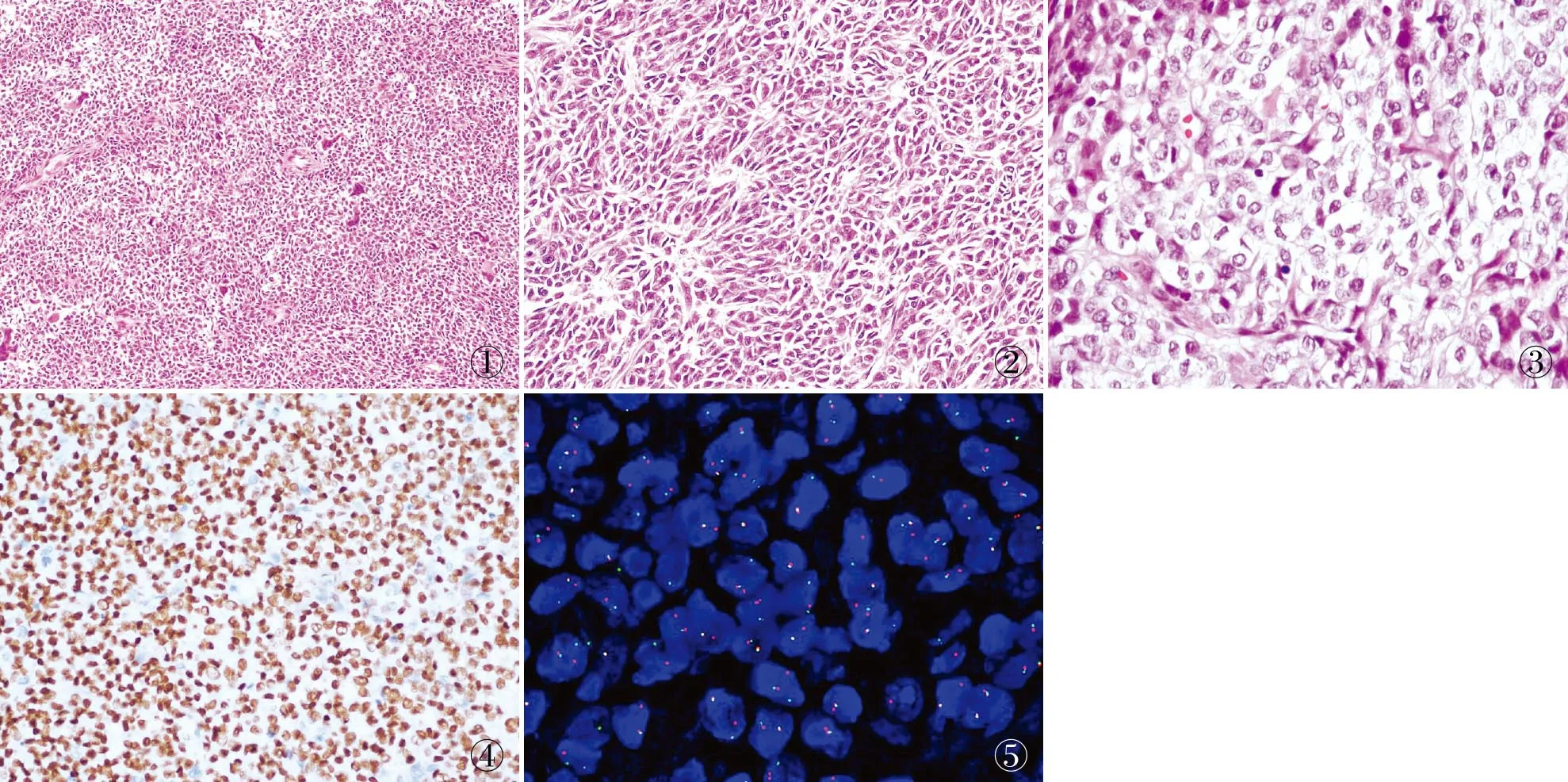
图1 肿瘤细胞呈实性片状排列,间质疏松水肿,可见散在分布的破骨样多核巨细胞
2.3 免疫表型瘤细胞弥漫表达SOX10(2/2,图4)、S-100蛋白(2/2),部分表达Syn(1/2)及CD56(2/2),局灶弱表达CD99(1/2),无H3K27Me3和INI1缺失,Ki67增殖指数为30%~70%,不表达CgA、HMB-45、Melan A、Fli-1、NKX2.2、desmin、MyoD1、Myogenin、CKpan、CD34、CD117、DOG1、PD-L1。
2.4 基因检测FISH检测2例均出现EWSR1基因分离信号(2/2,图5)。例1 NGS检测到EWSR1-CREB1融合基因(图6)和FANCL基因Exon10-11缺失突变,进一步行FISH基因检测重复验证,证实存在EWSR1-CREB1融合基因。
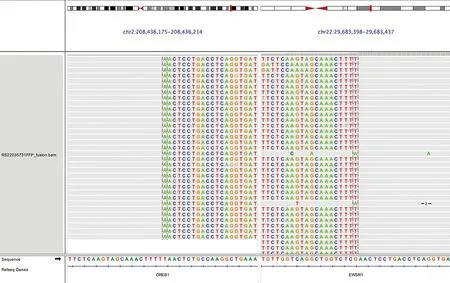
图6 NGS法检测示EWSR1-CREB1基因融合
2.5 预后2例均行手术切除+淋巴结清扫,例1术后予以表柔比星和异环磷酰胺辅助化疗4个疗程,随访7个月无复发及转移。例2术后辅以吉他西滨和多西他赛化疗,5个月后发现肝转移,并予以肝动脉栓塞治疗,2年后死亡(表1)。
3 讨论
3.1 概述2003年CCSLTGT/MGNET由Zambrano等[1]首次报道,2012年Stockman等[2]将其更名为MGNET,WHO(2019)消化系统肿瘤分类中将其命名为CCSLTGT/MGNET,其形态学和基因改变与软组织透明细胞肉瘤相似,但缺乏黑色素细胞分化的超微结构和免疫表型[3]。截至2022年5月,全球仅报道122例CCSLTGT/MGNET(包括本组2例),发病年龄5~82岁(中位年龄38岁),以女性略多见(男女比为0.83 ∶1),最常见发病部位为小肠(59.0%),其次是结肠(14.8%)、胃(13.9%)、食管(4.1%)及其他(8.2%)[4-8]。临床表现无特异性,多数患者表现为腹痛、肠梗阻。少数患者有厌食、体重减轻、贫血或发热等症状[4]。本组患者以腹痛、腹部肿块及大便习惯改变为主要症状。
3.2 诊断(1)眼观:肿瘤呈溃疡型或隆起型肿块,最大径为2.4~15 cm,平均为5.2 cm;中位最大径为3.8 cm[2]。切面质实,灰白色,伴不同程度出血和坏死。(2)镜检:肿瘤细胞呈实性片状、巢状排列、部分可呈假乳头状、菊形团样、假腺样、微囊性、束状及索状生长模式[4-5]。瘤细胞呈卵圆形、短梭形或上皮样,小-中等大,胞质嗜酸性或透明,核圆形,核仁一般不明显或可见小核仁,核分裂象平均10个/10 HPF[6],常有肿瘤性坏死,综合122例CCSLTGT/MGNET的特点,发现40.2%病例中可见散在的破骨样多核巨细胞。(3)免疫表型:肿瘤细胞表达S-100和SOX10,并不同程度表达CgA、Syn和CD56,不表达黑色素和胃肠间质瘤标志物[7-8]。(4)分子遗传学:具有典型的t(12;22)(q13;q12)与t(2;22)(q32.3;q12)易位产生EWSR1-ATF1和EWSR1-CREB1融合基因。最大宗两篇文献报道显示,EWSR1阳性率为86%(12/14)~93.3%(14/15)[2,4]。分析122例CCSLTGT/MGNET显示EWSR1阳性率为80.3%(分离及融合)。Kandler等[7]报道CCSLTGT/MGNET存在短片段基因改变,包括CDKN2A/B纯合性缺失、BCOR等短片段碱基替换、插入和缺失突变等。本组例1通过NGS检测发现CCSLTGT/MGNET存在EWSR1-CREB1融合基因和FANCL基因Exon10-11缺失突变。FANCL基因的缺失突变可能导致其后续编码框发生移码,进而导致编码的蛋白质表达缺失。此外,FANCL功能缺失可能导致同源重组修复缺陷,促进肿瘤对PARP抑制剂的敏感性[9]。本例扩展CCSLTGT/MGNET遗传学特征的认识,为后续靶向治疗提供依据。
3.3 鉴别诊断CCSLTGT/MGNET需与胃肠间质瘤、软组织透明细胞肉瘤、黑色素瘤、上皮样恶性周围神经鞘膜瘤等鉴别。(1)胃肠间质瘤:瘤细胞呈梭形,束状排列,免疫组化标记CD117、DOG1、CD34均阳性,分子检测有C-Kit或PDGFRα基因突变,可与之鉴别。(2)软组织透明细胞肉瘤:有黑色素沉积,免疫组化标记黑色素标志物阳性,电镜下见黑色素细胞分化的超微结构,支持软组织透明细胞肉瘤。(3)上皮样恶性周围神经鞘膜瘤:形态学和免疫表型有重叠,但分子检测无EWSR1基因重排有助于鉴别诊断。(4)黑色素瘤:瘤细胞异型性明显,形态学多样,可见核仁,细胞质和间质见不等量黑色素,免疫组化标记瘤细胞SOX10、S-100、HMB-45和Melan A均阳性,无H3K27Me3缺失;分子检测显示BRAF及C-Kit基因突变,可辅助鉴别诊断。
3.4 治疗目前,尚无针对CCSLTGT/MGNET标准化疗或靶向治疗方案,常见治疗方法是肿瘤根治切除并行淋巴结清扫。文献报道以异环磷酰胺和阿霉素为主的辅助化疗可使患者获益[7]。少数病例也有单独使用抗PD-L1抗体的免疫疗法,患者并未获益[4]。Kandler等[7]报道伴骨转移的CCSLTGT/MGNET患者,可以从放疗中获益。
3.5 预后CCSLTGT/MGNET患者总体预后差,生存期短,综合分析122例CCSLTGT/MGNET显示55.7%患者出现复发和转移。Li等[8]对有随访数据的76例患者回顾性分析发现,CCSLTGT/MGNET总生存期为0.69~161个月,中位时间为61个月;无瘤生存期为1~109个月,中位时间为10个月。本组1例术后随访7个月,患者无复发和转移,1例术后随访5个月发现肝转移,2年后死亡。
总之,CCSLTGT/MGNET是一种非常罕见的胃肠间叶源性肿瘤,临床表现无特异性,有相对特征性组织学、免疫表型及分子遗传学特征,患者预后差,生存期短。
