A robust genomic-based prognostic model for the assessment of cancer stemness and survival for patients with hepatocellular carcinoma
2024-03-04ChengLiDuShenYuWeiYunHoChenKngJieChen
Cheng-Li Du ,Shen-Yu Wei ,Yun-Ho Chen ,Kng-Jie Chen
a Division of Thoracic Surgery, First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou 310003, China
b Zhejiang University School of Medicine, Hangzhou 310003, China
c Division of Hepatobiliary Pancreatic Surgery, First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou 310003, China
TotheEditor:
Hepatocellular carcinoma (HCC) is an aggressive malignancy with poor long-term prognosis [1].Liver cancer stem cell (CSC) can drive the metastasis,drug resistance,and recurrence of HCC [2].The regulatory mechanisms about liver cancer stemness have been investigated,which encouraged the identification and characterization of novel prognostic and therapeutic strategies for HCC [3].As genome-wide,high-throughput sequencing became available,some prognostic models have been established based on gene expression profiles associated with HCC stemness [4].Nevertheless,in most cases,these models were hardly used in clinical practice due to complex algorithm and expensiveness.This study aimed to establish a stable stemness-related clinical prediction model with simplified algorithms based on gene expression profiles.Furthermore,biomolecular experiments were performed to examine the oncogenic functions of stemness-related genes.
A total of 903 HCC patients from four independent HCC cohorts [the Cancer Genome Atlas Liver Hepatocellular Carcinoma(TCGA-LIHC),International Cancer Genome Consortium Liver Cancer RIKEN Japan (ICGC-LIRI-JP),GSE14520,and GSE116174]were included.For TCGA-LIHC cohort,365 HCC samples and 50 normal liver tissue samples were included.The corresponding expression profiles,somatic mutation data and copy number variation (CNV) data,as well as detailed clinicopathological information,were obtained with the “TCGAbiolink” R package [5].And the standardized gene expression files and clinicopathological information of ICGC-LIRI-JP with 231 HCC samples and 202 normal liver tissues were obtained based on Illumina HiSeq RNA sequencing platform.The GSE14520 cohort included 242 HCC tissue samples,and the GSE116174 cohort included 65 HCC tissue samples,both of which contained complete clinical survival data.To assess the cancer stemness of patients in the aforementioned cohorts accurately,we used one-class logistic regression (OCLR) to calculate the stemness index based on mRNA expression (mRNAsi) of each patient based on expression matrix [6].The OCLR is a machine learning algorithm that trained on the datasets of stem cell classes and their differentiated ecto-,meso-and endoderm progenitors,and applied to RNA-seq datasets to calculate the mRNAsi based on transcriptomic and epigenetic signatures.The patients were divided into the high and low mRNAsi groups according to the median value of stemness indices.Kaplan-Meier survival analysis was used to evaluate the association of patient survival and mRNAsi.Next,genome-wide regions with significant amplification or deletion changes were identified by GISTIC 2.0,a biological program based on robust computational algorithms to detect somatic CNV by evaluating the frequency and magnitude of corresponding events [7].The somatic mutation annotation was analyzed in the high mRNAsi and low mRNAsi groups,and the significance of the top twenty genes was calculated with Wilcoxon test.The absolute number of genes with copy number changes in Focal and Arm levels is copy number loss load or copy number gain load [8].The copy number changes in Focal and Arm levels were compared between the high and low mRNAsi groups to reveal genomic instability in HCC patients with different cancer-stemness characteristics.
Subsequently,for the purpose of building a clinically practical prognostic model,the “Limma” R package was used for differential expression gene (DEG) analyses between the high and low mRNAsi groups under the threshold of | log2(fold change) |>0.5 and false discovery rate (FDR)<0.001.The intersection of DEGs in the two cohorts was defined as HCC stemness (HCS)-related genes.Then,we used univariate Cox analysis to screen and check prognostic HCS-related panel in HCC.TCGA-LIHC cohort was taken as a training set and risk model was constructed using Least absolute shrinkage and selection operator (LASSO) algorithm.The other three cohorts were used for model validation.Independent risk factors identified by multivariate Cox regression analysis were used to construct a nomogram to quantitatively predict the overall survival (OS) of HCC patients.Nomograms integrated multiple predictive indicators to score the outcome variables according to the scale corresponding to each predictive indicator,and finally evaluated the probability of outcome events of a patient according to the total score.Its construction and validation strictly follow the research guidelines of Lasonos et al.[9].
Finally,to investigate the biological functions of the cancer stemness-related hub genes,two HCC cell lines were purchased from the Liver Cancer Institute,Affiliated Zhongshan Hospital,Fudan University (Shanghai,China),including HCCLM3 and Huh7.Cell counting kit-8 (CCK8) assays and colony formation assays were carried out.
Based on mRNA expression matrix,OCLR algorithm was used to calculate the mRNAsi for each patient in the TCGA-LIHC cohort(n=365) and ICGC-LIRI cohort (n=231),respectively (Fig.1 A,B).The stemness index (mRNAsi) ranged from 0 to 1,and tumor samples were divided into the high and low mRNAsi groups based on the median value.The results showed that mRNAsi levels were significantly higher in HCC tumor tissues compared with those in adjacent tissues (P<0.001) (Fig.1 C).Next,we found that patients with higher mRNAsi levels had worse pathological grade(P<0.001) (Fig.1 D).Moreover,Kaplan-Meier analysis was performed,and patients in the TCGA-LIHC cohort with higher mRNAsi showed not only worse OS [hazard ratio (HR)=1.60,95%CI: 1.33-2.26,P=0.007],but also lower progression-free survival(PFS) (HR=1.76,95% CI: 1.26-2.44,P=0.002) and diseasefree survival (DFS) (HR=1.58,95% CI:1.18-2.13,P<0.001) (Fig.1 E-G).Similar results were observed in the ICGC-LIRI cohort (HR=4.24,95% CI: 2.31-7.78,P<0.001) (Fig.1 H).These results preliminarily suggested that the stemness of HCC was highly heterogeneous between normal and cancer tissues,and significantly affected the prognosis of HCC patients,so it may serve a pleuripotent role for HCC cancer cells to spread to extrahepatic metastases,tumor recurrence,as well as refractory drug resistance.
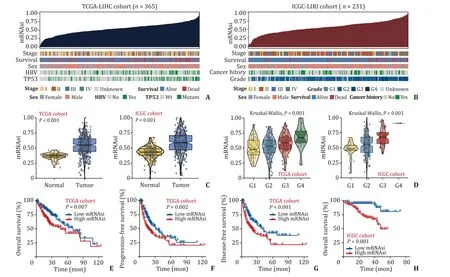
Fig.1. Association of cancer stemness index and clinicopathological features in HCC.A and B: Overview of mRNAsi and clinical features in the TCGA-LIHC and ICGC-LIRI cohorts.C: The mRNAsi was significantly higher in HCC than that in normal tissues.D: There was a significant positive correlation between mRNAsi and pathological grade of HCC.E-H: The relationship between mRNAsi and survival in HCC cohorts.HCC: hepatocellular carcinoma;TCGA-LIHC: The Cancer Genome Atlas Liver Hepatocellular Carcinoma;ICGC-LIRI: International Cancer Genome Consortium Liver Cancer RIKE.
Next,given the significant difference in survival benefit,we attempted to reveal the genomic instability between the high and low mRNAsi HCC groups.Somatic mutation annotation was analyzed,and the top 20 mutated genes in the high and low mRNAsi groups were identified,respectively (Fig.2 A,B).After comparison,a total of 23 genes with significant mutation differences were identified between the high and low mRNAsi groups (FDR<0.05)(Fig.2 C).In general,somatic mutations were more frequent in the high mRNAsi group (Fig.2 C).Notably,TP53 as a tumor suppressor gene,was mutated most frequently in patients with high mRNAsi (39% vs.18%,P<0.001).Likely,CNV was assessed.The GISTIC score and distribution of CNV frequency in chromosomes were demonstrated in Fig.2 D.It was found that at the Focal level,patients in the high mRNAsi group showed a higher copy number gain load (P<0.001) and loss load (P<0.001) (Fig.2 E).Whereas at the Arm level,patients in the high mRNAsi group had a higher copy number loss load (P<0.001) but not gain load (P=0.130)than those in the low mRNAsi group (Fig.2 F).These results imply a significant correlation between high stem cell characteristics and genomic instability in HCC,and high genomic variability may be one of the important reasons for poor prognosis in patients with high mRNAsi.
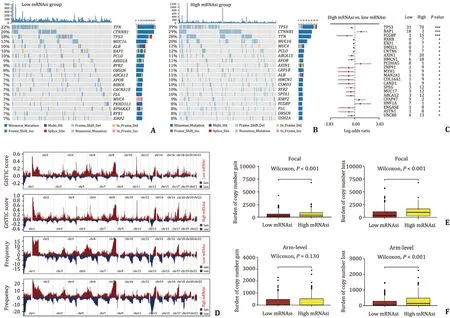
Fig.2. Association between the mRNAsi and genomic instability in HCC.A and B: Waterfall diagram shows the 20 most common mutated genes and their specific mutation forms in the high and low mRNAsi groups,respectively.C: Genes with the most significant mutation difference between the high and low mRNAsi groups.D: GISTIC score and variation frequency of copy number in autosomes in the high and low mRNAsi groups.E: At Focal level,difference of copy number gain and loss load between the high and low mRNAsi groups.F: At Arm level,difference of copy number gain and loss load between the high and low mRNAsi groups.∗: P < 0.05;∗∗: P < 0.01;∗∗∗: P < 0.001.ns: not significant;HCC: hepatocellular carcinoma.
Despite its prognostic value,mRNAsi is difficult to apply to clinical practice because it is algorithmically complicated and relies on whole-genome sequencing data.Hence,a simpler and more cost-effective prediction model that based on cancer stemness is urgently needed.Thereafter,to establish HCC specific tumor stemness prognostic model,we performed differential analysis in the TCGA-LIHC and ICGC-LIRI cohorts,respectively.A total of 569 mRNAsi-related DEGs were obtained from the intersection of two cohorts (Fig.3 A).Univariate Cox analysis was conducted,and a total of 92 prognostic DEGs were obtained,of which the vast majority were independent risk factors for HCC,and their expression levels were positively correlated with cancer stemness(data not shown).To identify genes with the most prognostic potential,we further performed LASSO regression analysis on these 92 genes (Fig.3 B).Seven HCS hub genes were identified:RAMP3,KLF2,TKT,NEIL3,CDCA8,TMEM106C,andKPNA2(Fig.3 C).HCC stemness-related risk score (HCSRS) model was constructed based on expression level of hub genes and their corresponding regression coefficient: HCSRS=0.223×KPNA2+0.062×CDCA8-0.123×RAMP3+0.057×NEIL3+0.075×TMEM106C-0.013×KLF2+0.047×TKT.Based on this formula,the HCSRS of each patient in the TCGA-LIHC training cohort was calculated,and patients with high HCSRS had worse OS compared with those with low HCSRS (HR=2.66,95% CI: 1.87-3.76,P<0.001) (Fig.3 D).ROC curve was used to evaluate the clinical prediction performance of HCSRS.The AUC values of the HCSRS model in predicting 1-,2-and 3-year OS were 80%,75% and 75%,respectively in the training cohort (Fig.3 E),indicating that the HCSRS model has a fair clinical predictive performance.Moreover,three independent validation cohorts including ICGC-LIRI,GSE14520 and GSE116174,were used to evaluate the stability of HCSRS prognostic performance.In line with the training cohort,the HCSRS still possessed well clinical significance for outcome prediction (P<0.05) (Fig.3 D).And ROC curve also proved that HCSRS had high predictive accuracy for 1-,2-and 3-year OS in the validation cohorts (Fig.3 E).Multivariate Cox analysis further suggested that the HCSRS was an independent risk factor of poor prognosis for HCC patients (Fig.3 F),and confirmed its robust predictive efficiency (HR=4.50,95% CI:2.57-7.88,P<0.001).The Sankey diagram showed that this prediction model could be better applied in assessing the tumor stemness of patients individually,that is,patients with high risk score had higher stemness index,which would mean higherp53mutation and CNV,and worse clinical outcome (Fig.3 G).In addition,compared with OCLR-based prediction algorithm,our model was more concise and had more clinical application value.
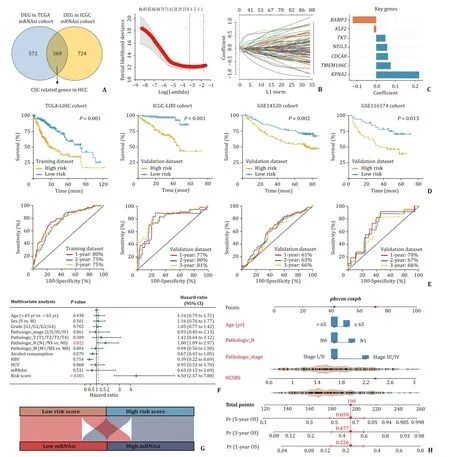
Fig.3. Establishment of a nomogram integrating HCC stemness-related risk model.A: Differentially expressed genes between the high and low mRNAsi groups in TCGA and ICGC cohorts.B: LASSO regression identifies seven of the most potential mRNAsi-related prognostic genes,and 10×cross-validation.C: Distribution of regression coefficients of mRNAsi-related prognostic hub genes.D: TCGA-LIHC (training set) was used to construct the HCC stemness-related risk score (HCSRS) model,and the patients with high HCSRS suffered worse OS than those with low HCSRS.ICGC-LIRI,GSE14520 and GSE116174 function as external validation sets to evaluate the performance of the risk model,and the OS of high-risk patients is statistically worse.E: ROC curve was used to evaluate the sensitivity and specificity of HCSRS in predicting patient OS in the training set and validation set.F: Multivariate Cox analysis revealed that HCSRS is an independent risk factor influencing the prognosis of HCC patients.G: Sankey diagram showed that patients with high risk score possessed high mRNAsi.H: Nomogram was constructed to quantitatively predict the death risk of HCC patients at 1,3 and 5 years.HCC:hepatocellular carcinoma;HCSRS: HCC stemness-related risk score;OS: overall survival;ROC: receiver operating characteristic.LASSO: Least absolute shrinkage and selection operator;TCGA-LIHC: The Cancer Genome Atlas Liver Hepatocellular Carcinoma;ICGC-LIRI: International Cancer Genome Consortium Liver Cancer RIK.
To better quantify and materialize our clinical stemness model,two independent prognostic factors (N stage and HCSRS) in the TCGA cohort were integrated and a nomogram was established combining patient age and TNM stage (Fig.3 H).The C-index of the established nomogram was 0.75 (95% CI: 0.71-0.78) after 1000 bootstrap iterations.The calibration plots also proved that the nomogram performed good consistency when compared predicted survival with actually observed outcomes at 1,3 and 5 years,indicating that the nomogram had strong,robust and quantitative clinical prognostic ability for the hazard classification of HCC patients.
Additionally,the expression of hub genes at the protein levels were validated in HCC tumor tissues and normal liver tissues.Most of oncogenes in our model showed significant higher expression in HCC tissues compared with that in normal liver tissues by immunohistochemistry (Fig.4 A).KPNA2,one of the hub genes,was chosen for validation with the heaviest weight in our model.KPNA2 was highly expressed in the HCC tumor tissues (Fig.4 B)and its high expression was associated with worse OS (Fig.4 C).Next,KPNA2 was silenced by siRNA (Fig.4 D) to investigate its biological roles (in terms of proliferation) in HCCLM3 cell lines.CCK8 and colony formation assays were carried out,and the results showed that the proliferation of HCC cells were significantly reduced upon KPNA2 silencing (Fig.4 E,F).These results suggested that the stemness-related hub genes promoted HCC progression with through different manners.
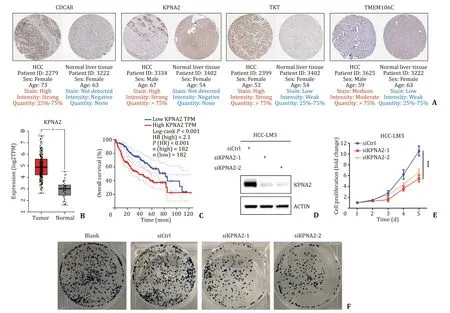
Fig.4. Experimental validation of expression and functions of hub genes.A: Expression levels of HCC cancer stemness-related hub genes at protein level between tumor and normal tissues were analyzed based on immunohistochemical.B: KPNA2 was differentially expressed in tumor and normal tissues at the mRNA level.C: Kaplan-Meier survival analysis showing the overall survival with high-and low-KPNA2 expression.D: Knockdown efficiency of KPNA2 was validated by Western blot.E and F: Cell counting kit-8 assays and cell colony formation assay to observe the proliferation of HCC cells after siRNA transfection.∗: P < 0.05;∗∗: P < 0.01;∗∗∗: P < 0.001;ns: not significant;HCC: hepatocellular carcinoma.
Overall,through the multidisciplinary approach of computer science and life science,and based on the large-scale HCC research cohort,this study comprehensively and systematically analyzed the potential relationship between the characteristics of HCC stem cells and genetics,clinical prognostic characteristics and biomolecular pathways.Seven HCC stemness-related hub genes were screened out,and a more efficient and high clinical application value stemness-related clinical prediction model was constructed,which can quantitatively predict the mortality risk of patients,identify tumor heterogeneity and evaluate patients’ individual cancer stemness level.These results will provide a promising theoretical basis for the individualized treatment of HCC patients from the perspective of cancer stemness.
Acknowledgments
None.
CRediT authorship contribution statement
Cheng-Li Du:Data curation,Funding acquisition,Writing–original draft.Shen-Yu Wei:Methodology,Formal analysis.Yun-Hao Chen:Writing– original draft.Kang-Jie Chen:Conceptualization,Supervision,Writing– review &editing.
Funding
This study was supported by a grant from the National Natural Science Foundation of China (82002927).
Ethical approval
Not needed.
Competing interest
No benefits in any form have been received or will be received from a commercial party related directly or indirectly to the subject of this article.
杂志排行

Hepatobiliary & Pancreatic Diseases International的其它文章
- Recent advances in promising drugs for primary prevention of gastroesophageal variceal bleeding with cirrhotic portal hypertension
- Stereotactic body radiotherapy in pancreatic adenocarcinoma
- Application of ultrasonography-elastography score to suspect porto-sinusoidal vascular disease in patients with portal vein thrombosis
- Polydatin ameliorates hepatic ischemia-reperfusion injury by modulating macrophage polarization
- Hypomethylation of glycine dehydrogenase promoter in peripheral blood mononuclear cells is a new diagnostic marker of hepatitis B virus-associated hepatocellular carcinoma
- AGK2 pre-treatment protects against thioacetamide-induced acute liver failure via regulating the MFN2-PERK axis and ferroptosis signaling pathway