经典名方葛根芩连汤基准样品的HPLC 指纹图谱及量质传递规律研究
2024-03-02陈佳美成颜芬杨晓琴傅超美章津铭吴亿晗
陈佳美,陈 蓉,成颜芬,杨晓琴,王 潇,傅超美,章津铭,吴亿晗
成都中医药大学药学院,西南特色中药资源国家重点实验室,中药材标准化教育部重点实验室,四川 成都 611137
经典名方葛根芩连汤( Gegen Qinlian Decoction,GQD)出自东汉著名医家张仲景所著《伤寒论》第34 条,原方名为葛根黄芩黄连汤,至近代《中国医药大辞典》始成“葛根芩连汤”方名[1]。全方由葛根、黄芩、黄连、炙甘草4 味中药组成,方中以葛根为君,既能清热解表,又能升发脾胃清阳之气而治下痢;臣药以黄芩、黄连苦寒之品可清胃肠之湿,使以炙甘草清热解毒,调和诸药[2]。该方具有表里双解、清热止利之功,为治热陷阳明、协热下利常用方[3]。现代研究表明,GQD 具有止泻、抗炎抑菌、解热、抗心律失常、降糖等药理作用[4]。可用于治疗胃肠型感冒、细菌性痢疾、糖尿病、高血压病、高脂血症、溃疡性结肠炎等疾病[5]。
《中药注册分类及申报资料要求》[6]将第3 类古代经典名方中药复方制剂细分为“3.1 按古代经典名方目录管理的中药复方制剂”(以下简称“3.1 类”)及“3.2 其他来源于古代经典名方的中药复方制剂”(以下简称“3.2 类”)。在“3.1 类”经典名方相关政策出台逐步完善、其制剂的关键技术研发讨论热这一趋势下,本研究参考“3.1 类”经方制剂研发思路开发“3.2 类”经典名方GQD 具有巨大潜力与研究意义。在《古代经典名方中药复方制剂基准样品的申报资料要求(征求意见稿)》中提出了为保障制剂的质量,应重点关注基准样品质量控制体系的建立,该质量控制体系的建立应以按照国家发布的古代经典名方关键信息及古籍记载内容制备的基准样品为前提[7],以出膏率、含量测定、指纹图谱或特征图谱等为指标,进行量质传递分析,从而保证基准样品质量的均一性和可追溯性[8]。
目前,对于GQD 中4 味中药的化学基础及主要活性成分的研究较为清楚[9-13],其相关制剂的整体质量控制的研究,多为以HPLC 同时测定多种有效成分为主,其中,章军等[9]、李丽莉等[14]、毛莹等[15]分别建立了葛根芩连相关制剂中10 种以上有效成分,在不同波长下同时测定的HPLC 含量测定方法,胡晓茹等[16]对葛根芩连片特征图谱中8 个特征峰进行指认。因此,本研究在前期文献考证明确《伤寒论》中记载的GQD 的饮片处方量、加水量、煎煮时间、煎煮次数的基础上,拟制备15 批GQD基准样品,并分别建立了4 味中药中关键指标成分的含量测定方法,优化前期研究中葛根芩连相关制剂的HPLC 指纹图谱条件,以出膏率、含量测定、指纹图谱或特征图谱等为指标,探明GQD 指标成分在“饮片-水煎液-冻干粉”的传递规律,为后续GQD 经方制剂的质量控制奠定了研究基础。
1 仪器与试药
1.1 仪器
UPK-I-107 型优普超纯水器,四川优普超纯科技有限公司;FTS-10A 型全自动煎药壶,潮州市一壶百饮电器实业有限公司;Sartorius CPA225D 型电子天平,十万分之一,赛多利斯科学仪器(北京)有限公司;HTP-312 型电子天平,万分之一,上海华潮电器有限公司;SCIENTZ-10N 型冷冻干燥机,宁波新芝生物科技有限公司;DL-720D 型超声机,上海之信仪器有限公司;UltiMate 3000 型高效液相色谱仪,美国Thermo Fisher 公司;色谱柱Xterra RP18(250 mm×4.6 mm,5μm),沃特世科技(上海)有限公司;色谱柱5C18-MS-II(250 mm×4.6 mm,5 μm),上海屹利科学仪器有限公司;SHZ-D(III)型循环水式多用真空泵,巩义市予华仪器有限公司;N-1100 型旋转蒸发仪,上海泉杰仪器有限公司;Scientz-10N 型冷冻干燥机,宁波新芝生物科技有限公司。
1.2 试药
对照品葛根素(批号wkq22012601)、大豆苷(批号wkq22041304)、大豆苷元(批号wkq16080304)、黄芩苷(批号 wkq21010608)、黄芩素(批号wkq21030203)、汉黄芩苷(批号wkq21031811)、汉黄芩素(批号wkq21022605)、黄连碱(批号wkq21030107)、盐酸小檗碱(批号wkq21022201)、巴马汀(批号wkq21050608)、表小檗碱(批号wkq22032907)、甘草苷(批号wkq18040902)、甘草酸铵(批号wkq22011005)均购于成都维克奇生物科技有限公司,各对照品质量分数均≥98%。水为超纯水;乙腈(色谱级,湖北弗顿科学技术有限公司、磷酸、甲醇(色谱级,成都市诺尔施科技有限责任公司),西格玛奥德里奇(上海)贸易有限公司。葛根、黄芩、黄连、甘草药材均购自四川新荷花中药饮片股份有限公司,经成都中医药大学蒋桂华教授鉴定,分别为豆科葛属植物野葛Pueraria lobata(Willd.) Ohwi 的干燥根、唇形科黄芩属植物黄芩ScutellariabaicalensisGeorgi 的干燥根、毛茛科黄连属植物黄连CoptischinensisFranch.的干燥根茎、豆科甘草属植物甘草GlycyrrhizauralensisFisch.的干燥根和根茎。葛根、黄芩、黄连、炙甘草饮片均为自制[17],药材产地与饮片炮制加工方法见表1。
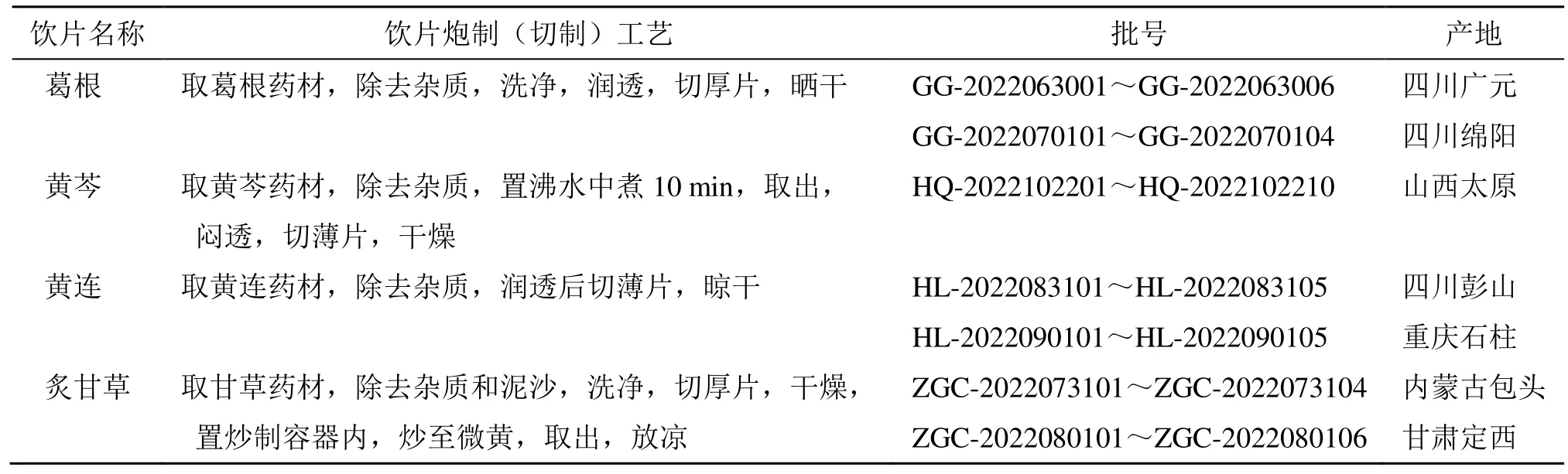
表1 GQD 饮片来源Table 1 Sources of GQD pieces
2 方法与结果
2.1 15 批GQD 基准样品的制备
2.1.1 药材基原、饮片炮制、用法用量考证 参照《中国药典》2020 年版一部[17],明确了葛根为豆科葛属植物野葛P.lobata(Willd.) Ohwi 的干燥根、黄芩为唇形科黄芩属植物黄芩S.baicalensisGeorgi 的干燥根。根据《中国药典》2020 年版以及国家中医药管理局、国家药品监督管理局联合发布的32 首经典名方关键信息[18]中来源于《伤寒论》的经方对于黄连的基原记载,明确了黄连为多基原植物,其来源于毛茛科黄连属植物黄连C.chinensisFranch.、三角叶黄连C.deltoideaC. Y. Cheng et Hsiao 或云连C.teetaWall.的干燥根茎。在本研究中考虑到实验的时效性以及药材方便购买程度,固定了黄连基原为毛茛科黄连属植物黄连C.chinensisFranch.的干燥根茎做进一步研究。经课题组前期对于甘草的本草考证发现,所记载的甘草基原均为乌拉尔甘草G.uralensisFisch.,未见药典所载胀果甘草G.inflataBat.和光果甘草G.glabraL.[17,19-22]。参照32 首经典名方关键信息来源于《伤寒论》的经方对于甘草基原记载,其明确规定了甘草药材基原为豆科甘草属植物甘草G.uralensisFisch.的干燥根和根茎。对于甘草的“炙”法,药典规定为蜜炙,然而蜜作为辅料最早出现于唐代,在此之前未提及关于辅料的记载,参照32 首经典名方关键信息,此处的“炙”法应为清炒法[17,22]。
对于处方剂量,原文记载:“葛根(半斤)、黄芩(三两)、黄连(三两)、甘草(二两)……,上四味,以水八升,先煮葛根,减二升,内诸药,煮取二升,去滓……”[18]。参照古代计量史专家丘光明、邱隆、杨平合著,由科技出版社出版的《中国科学技术史·度量衡卷》[23],明确东汉时期的1 两折合为13.8 g,1 升折合成现今200 mL,“去滓”多指过滤除去药渣[24],由此可知,《伤寒论》中GQD 的制法为加水1 600 mL,先煮葛根(110.4 g),当煮至汤液体积为1 200 mL,再加入黄芩(41.4 g)、黄连(41.4 g)、炒甘草(27.6 g),煮至汤液为400 mL,过滤除去药渣。
2.1.2 15 批GQD 基准样品的制备 采用随机数表法对4 味饮片进行随机组合及排序,组成15 批GQD基准样品对应饮片的批号。按前期研究得到的方法制备GQD 基准样品,先将葛根置于紫砂壶中,加入1 600 mL 纯水,设定浸泡时间为30 min,参考传统武火煮沸、文火保持微沸的汤液煎煮方法,武火煮沸后,转文火煎煮至汤液体积为1 200 mL 时,加入黄芩、黄连、炙甘草,煮至汤液为400 mL。150目无纺布趁热滤过,滤液减压浓缩至一定体积,得葛根芩连水煎浓缩液,将浓缩液置入冷冻干燥机中进行冷冻干燥,即得到15 批GQD 基准样品。
2.2 GQD 基准样品指纹图谱的建立
2.2.1 色谱条件 色谱柱为5C18-MS-II(250 mm×4.6 mm,5 μm);流动相为乙腈-0.02 mol/L 醋酸铵加入0.03%三乙胺(用冰醋酸调节pH 值为4.3),梯度洗脱:0~5 min,15%乙腈;5~10 min,15%~20%乙腈;10~15 min,20%~25%乙腈;15~30 min,25%乙腈;30~33 min,25%~40%乙腈;33~48 min,40%~50%乙腈;48~57 min,50%~65%乙腈;57~60 min,65%~15%乙腈;60~65 min,15%乙腈;体积流量1.0 mL/min;检测波长为280 nm;柱温30 ℃;进样量10 µL。
2.2.2 GQD 基准样品供试品溶液的制备 取GQD基准样品0.2 g 于25 mL 棕色量瓶中,精密加70%甲醇25 mL,超声(250 W、40 kHz)30 min,取出,待冷却后加70%甲醇定容至刻度,摇匀,转移至50 mL 离心管中,1 000 r/min 离心10 min(离心半径16 cm),取上清液过0.45 µm 微孔滤膜,即得。
2.2.3 单味饮片和阴性对照供试品溶液的制备 分别称取处方量的葛根、黄芩、黄连、炙甘草4 味饮片,按“2.1”项下GQD 基准样品制备工艺和“2.2.2”项下的处理方法,制得单味饮片供试品溶液;同法按处方量分别制备缺葛根、缺黄芩、缺黄连、缺甘草的4 个阴性对照供试品溶液。
2.2.4 对照品溶液的制备 分别取葛根素、大豆苷、大豆苷元、黄芩苷、黄芩素、汉黄芩苷、汉黄芩素、黄连碱、盐酸小檗碱、巴马汀、表小檗碱、甘草苷、甘草酸铵适量,精密称定,用70%甲醇分别制成含葛根素1.013 mg/mL、大豆苷1.005 mg/mL、大豆苷元0.995 mg/mL、黄芩苷2.120 mg/mL、黄芩素1.020 mg/mL、汉黄芩苷0.520 mg/mL、汉黄芩素1.067 mg/mL、黄连碱1.005 mg/mL、盐酸小檗碱1.045 mg/mL、巴马汀1.020 mg/mL、表小檗碱0.990 mg/mL、甘草苷0.995 mg/mL、甘草酸铵1.030 mg/mL 的对照品储备液。
2.2.5 精密度考察 取GQD 基准样品冻干粉,按“2.2.2”项下方法制备同一批GQD 基准样品冻干粉(S6)供试品溶液,按“2.2.1”项下色谱条件连续进样6 次,结果指纹图谱相似度>0.96;以6 号峰为参照峰,计算各共有峰相对保留时间和相对峰面积的RSD 均<2%,表明仪器精密度良好。
2.2.6 重复性考察 按“2.2.2”项下方法平行制备同一批GQD 基准样品冻干粉(S6)的供试品溶液6份,按“2.2.1”项下色谱条件进样分析,结果指纹图谱相似度>0.98;以6 号峰为参照峰,计算各共有峰相对保留时间和相对峰面积的RSD 均<2%,表明方法的重复性良好。
2.2.7 稳定性考察 按“2.2.2”项下方法制备GQD基准样品冻干粉(S6)供试品溶液1 份,按“2.2.1”项下色谱条件,分别于样品制备后的0、12、15、18、24、30 h 后进样分析,结果指纹图谱相似度>0.96;以6 号峰为参照峰,计算各共有峰相对保留时间和相对峰面积的RSD 均<2%,表明本方法的稳定性良好。
2.2.8 化学指纹图谱的建立及其相似度评价 根据“2.2.1”项下色谱条件对15 批次GQD 基准样品供试品溶液进行检测,将样品所得色谱数据导入中药色谱指纹图谱相似度评价系统(2012A 版)软件进行分析,利用中位数法,时间窗宽度设置为0.2 min,以S6 为对照,生成HPLC 叠加指纹图谱和对照指纹图谱,结果见图1。共标定了34 个共有峰,对15批GQD 基准样品相似度进行计算,得到不同批次的指纹图谱的相似度均>0.993,符合指纹图谱要求,结果见表2。

图1 15 批GQD 基准样品指纹图谱和对照指纹图谱的HPLC 图Fig. 1 Fingerprints of 15 batches of GQD benchmark samples and HPLC of control fingerprints
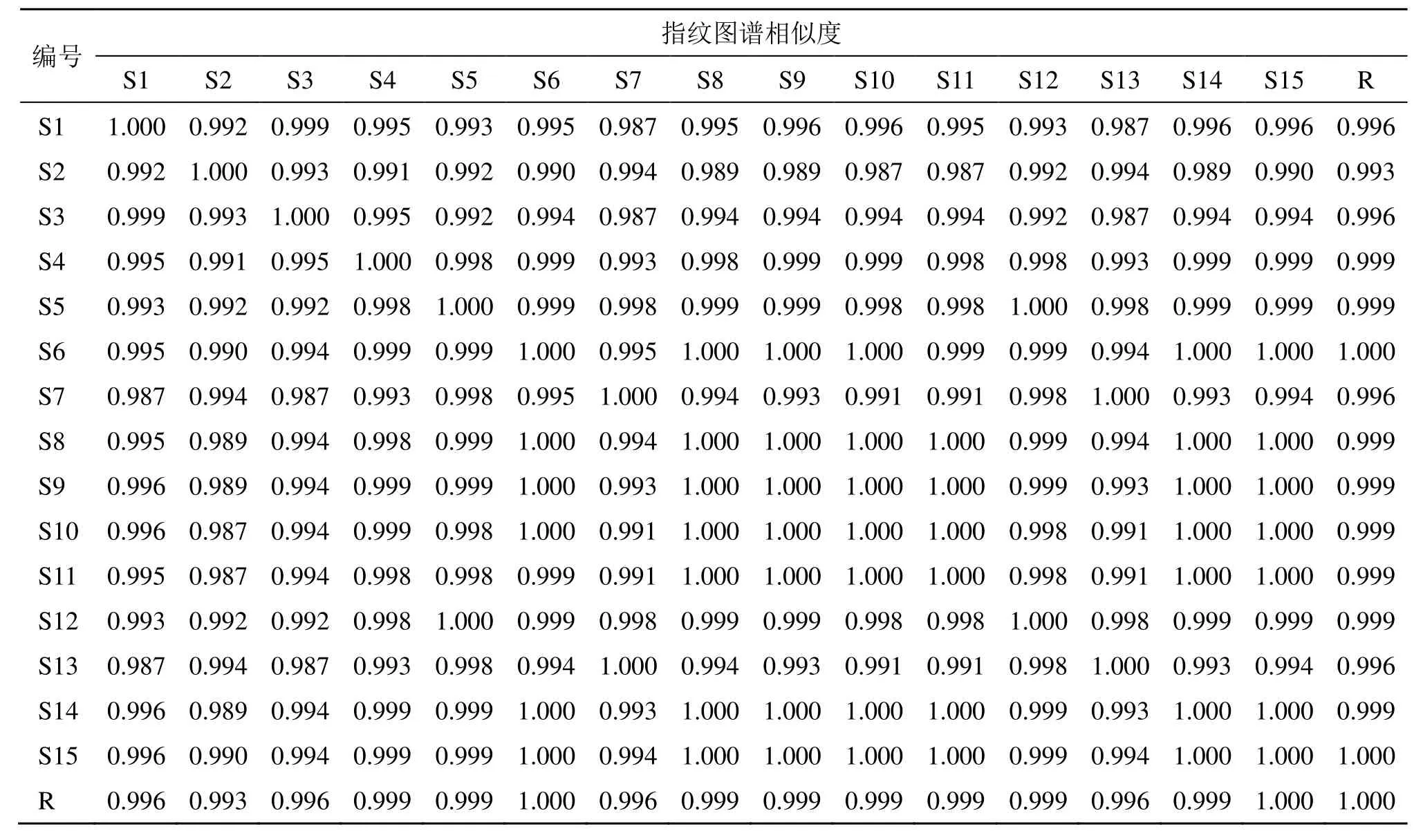
表2 15 批GQD 基准样品指纹图谱相似度评价结果Table 2 Similarity evaluation results of 15 batches of GQD benchmark samples
经与化学对照品的色谱行为进行比较,指认了13 个共有峰,分别为葛根素(6 号峰)、大豆苷(10号峰)、甘草苷(15 号峰)、黄芩苷(20 号峰)、表小檗碱(24 号峰)、黄连碱(25 号峰)、汉黄芩苷(26号峰)、大豆苷元(28 号峰)、巴马汀(29 号峰)、盐酸小檗碱(30 号峰)、甘草酸铵(32 号峰)、黄芩素(33 号峰)、汉黄芩素(34 号峰)。对照品溶液、供试品溶液、空白溶剂的HPLC 图见图2。

图2 空白溶剂 (A)、混和对照品 (B) 和GQD 基准样品(C) 的HPLC 图Fig. 2 HPLC of solvent blank (A), mixed reference substances (B) and GQD benchmark sample (C)
2.2.9 指纹图谱量质传递关系分析 通过分别将4种单味饮片样品、缺单味饮片阴性样品和全方基准样品指纹图谱进行比对,对标定的34 个共有峰进行归属,结果见图3。结果表明,4、6(葛根素)、7、9、10(大豆苷)、12、28(大豆苷元)号峰来源于葛根,11、14、16、19、20(黄芩苷)、21、22、26(汉黄芩苷)、27、33(黄芩素)、34(汉黄芩素)来源于黄芩,3、8、13、23、24(表小檗碱)、25(黄连碱)、29(巴马汀)、30(盐酸小檗碱)来源于黄连;15(甘草苷)、29、31、32(甘草酸铵)来源于甘草,5 号峰在葛根、黄连均有出现,17、18 号峰在葛根、黄芩、黄连均有出现,1、2 号峰在4 味中药中均有出现,推测可能为极性相似的一类化合物,有待进一步研究。

图3 GQD 基准样品指纹图谱 (R) 中34 个共有峰及其归属Fig. 3 Common peaks and their assignments in fingerprint chromatogram of GQD benchmark sample (R)
2.3 GQD基准样品中各指标成分含量在饮片-水煎液-基准样品中的传递规律研究
2.3.1 HPLC 色谱条件
(1)葛根饮片、GQD 水煎液、GQD 基准样品中葛根素、大豆苷、大豆苷元:5C18-MS-II 色谱柱(250 mm×4.6 mm,5 μm);流动相为甲醇-水,梯度洗脱:0~20 min,25%甲醇;20~30 min,25%~45%甲醇;30~40 min,45%~60%甲醇;40~50 min,60%~75%甲醇;体积流量1.0 mL/min;检测波长250 nm;柱温35 ℃;进样量10 µL。
(2)黄芩饮片、GQD 水煎液、GQD 基准样品中黄芩苷、黄芩素、汉黄芩苷、汉黄芩素:Xterra RP18 色谱柱(250 mm×4.6 mm,5 μm);流动相为乙腈-0.1%甲酸水溶液,梯度洗脱:0~10 min,25%~30%乙腈;10~30 min,30%~50%乙腈;30~35 min,50%~100%乙腈;35~40 min,100%~25%乙腈;体积流量1.0 mL/min;检测波长275 nm;柱温30 ℃;进样量10 µL。
(3)甘草饮片、GQD 水煎液、GQD 基准样品中甘草苷、甘草酸铵:Xterra RP18 色谱柱(250 mm×4.6 mm,5 μm);流动相为乙腈-0.1%磷酸水溶液,梯度洗脱:0~3 min,19%乙腈;3~20 min,19%~26%乙腈;20~22 min,26%~40%乙腈;22~34 min,40%乙腈;34~36 min,40%~19%乙腈;体积流量0.7 mL/min;检测波长237 nm;柱温35 ℃;进样量10 µL。
(4)黄连饮片、GQD 水煎液、GQD 基准样品中盐酸小檗碱、巴马汀:Xterra RP18 色谱柱(250 mm×4.6 mm,5 μm);流动相为乙腈-0.05 mol/L 磷酸二氢钾水溶液(用磷酸调节pH 值为3.0)25∶75;体积流量1 mL/min;检测波长346 nm;柱温30 ℃;进样量10 µL。
2.3.2 水煎液供试品溶液的制备 精密吸取水煎液0.2 mL 于5 mL 棕色量瓶中,加入70%甲醇2 mL,1 000 r/min 离心10 min(离心半径16 cm),取上清液过0.45 µm 微孔滤膜,即得。
2.3.3 基准样品供试品溶液的制备 同“2.2.2”项下。
2.3.4 单味饮片供试品溶液的制备 同“2.2.3”项下。
2.3.5 阴性对照供试品溶液的制备 同“2.2.3”项下。
2.3.6 对照品溶液的制备 按“2.2.4”项下方法配制的不同质量浓度的葛根素、大豆苷、大豆苷元、黄芩苷、汉黄芩苷、黄芩素、汉黄芩素、盐酸小檗碱、巴马汀、甘草苷、甘草酸铵11 个对照品溶液。
2.3.7 系统适用性考察 取上述基准样品、单味饮片、阴性对照供试品溶液,以及对照品溶液,按“2.3.1”项下各HPLC 色谱条件分别进行测定,记录色谱图,结果各成分分离度均>1.5,符合《中国药典》2020 年版要求,峰形较好,理论塔板数(n)以保留时间(tR)和峰宽(W)按n=16(tR/W)2计算均不低于15 000,阴性样品无干扰。
2.3.8 线性关系考察 将“2.3.6”项下对照品溶液按比例稀释2 倍形成系列梯度质量浓度,各指标成分按“2.3.1”项下各HPLC 色谱条件进样分析,以峰面积为横坐标(X)、以质量浓度为纵坐标(Y)进行线性回归方程计算,各指标成分的回归方程、相关系数(r)及线性范围分别为葛根素Y=0.558 0X+1.456 3,R2=0.999 9,线性范围3.96~1 012.50µg/mL;大豆苷Y=0.653 6X+2.109 0,R2=0.999 7,线性范围1.96~502.50 µg/mL;大豆苷元Y=1.258 7X+2.367 8,R2=0.999 7,线性范围1.94~497.50µg/mL;黄芩苷Y=0.614 8X-3.939 7,R2=1.000 0,线性范围16.56~2 120.00 µg/mL;汉黄芩苷Y=0.556 6X-1.531 9,R2=0.999 4,线性范围2.03~520.00 µg/mL;黄芩素Y=0.856 4X+0.018 5,R2=1.000 0,线性范围1.99~510.00 µg/mL;汉黄芩素Y=0.940 1X+1.888 9,R2=0.999 7,线性范围2.07~530.00 µg/mL;盐酸小檗碱Y=0.403 3X+0.054 1,R2=1.000 0,线性范围4.10~523.30 µg/mL;巴马汀Y=0.403 5X+0.023 6,R2=1.000 0,线性范围4.0~502.20 µg/mL;甘草苷Y=0.449 7X+0.683 7,R2=0.999 9,线性范围3.87~495.00 µg/mL;甘草酸铵Y=0.113 4X-0.967 4,R2=0.999 3,线性范围4.02~515.00 µg/mL。
2.3.9 精密度考察 取编号为S13 的GQD 基准样品,按“2.3.3”项下制备其供试品溶液1 份,参照“2.3.1”项下4 种HPLC 色谱条件,分别连续进样6次,记录峰面积,结果显示,11 种指标成分葛根素、大豆苷、大豆苷元、黄芩苷、汉黄芩苷、黄芩素、汉黄芩素、盐酸小檗碱、巴马汀、甘草苷、甘草酸铵峰面积的RSD 分别为1.7%、0.5%、0.8%、1.2%、1.5%、1.0%、0.3%、0.8%、0.9%、1.3%、0.7%,均小于2.0%,表明仪器精密度良好。
2.3.10 稳定性考察 取编号为S13 的GQD 基准样品,按“2.3.3”项下制备其供试品溶液1 份,参照“2.3.1”项下4 种HPLC 色谱条件,分别在制备后 0、2、4、8、12、24 h 进样,结果显示,11 种指标成分葛根素、大豆苷、大豆苷元、黄芩苷、汉黄芩苷、黄芩素、汉黄芩素、盐酸小檗碱、巴马汀、甘草苷、甘草酸铵峰面积的RSD 分别为1.9%、0.8%、0.6%、1.2%、1.8%、0.3%、1.0%、1.1%、0.7%、1.2%、1.0%,均小于2.0%,表明基准样品供试品溶液在24 h 内基本稳定。
2.3.11 重复性考察 取编号为S13 的GQD 基准样品,按“2.3.3”项下方法平行制备6 份其供试品溶液,参照“2.3.1”项下4 种HPLC 色谱条件,分别进样分析,结果显示,11 种指标成分葛根素、大豆苷、大豆苷元、黄芩苷、汉黄芩苷、黄芩素、汉黄芩素、盐酸小檗碱、巴马汀、甘草苷、甘草酸铵峰面积的RSD 分别为0.9%、1.6%、1.3%、0.4%、0.9%、1.2%、1.8%、0.5%、1.5%、0.7%、1.2%,均小于2.0%,表明该方法重复性良好。
2.3.12 加样回收率考察 精密称取编号为S13 的已知指标成分含量的GQD基准样品2.00 g于25 mL量瓶中,分别按样品中各成分含量的100%水平加入“2.3.6”项下11 种对照品溶液,按“2.3.3”项下方法平行制备6 份其供试品溶液,参照“2.3.1”项下4 种HPLC 色谱条件,分别进样分析,进行含量测定,计算11 种指标成分的加样回收率及RSD 值,结果葛根素、大豆苷、大豆苷元、黄芩苷、汉黄芩苷、黄芩素、汉黄芩素、盐酸小檗碱、巴马汀、甘草苷、甘草酸铵的平均加样回收率分别为102.79%、98.89%、103.25%、104.22%、99.78%、105.10%、96.87%、99.10%、101.76%、101.44%、99.76%,RSD 分别为0.6%、0.7%、0.6%、0.8%、1.0%、1.2%、1.1%、0.9%、1.2%、1.0%、0.9%,表明该方法加样回收率良好。
2.3.13 GQD 处方各味药指标成分在饮片、水煎液、基准样品中的含量测定结果 按“2.3.1”项下方法分别测定15 批饮片、水煎液、基准样品中11 个指标成分的含量,结果见表3~5。

表3 饮片中指标性成分的含量Table 3 Content of index components in decoction pieces

表4 水煎液中指标性成分的含量Table 4 Content of index components in decoction

表5 基准样品中指标性成分的含量Table 5 Content of index components in benchmark samples
15 批饮片中各成分质量分数分别为葛根素92.142~116.619 mg/g、大豆苷20.412~28.744 mg/g、大豆苷元1.338~1.989 mg/g、黄芩苷150.442~162.818 mg/g、汉黄芩苷48.887~51.232 mg/g、黄芩素4.419~4.818 mg/g、汉黄芩素1.848~2.485 mg/g、盐酸小檗碱66.437~88.617 mg/g、巴马汀17.446~22.016 mg/g、甘草苷19.950~20.613 mg/g、甘草酸铵43.400~52.267 mg/g;15 批水煎液中各成分质量分数分别为葛根素8.891~11.205 mg/g、大豆苷1.206~1.949 mg/g、大豆苷元0.162~0.260 mg/g、黄芩苷6.577~7.285 mg/g、汉黄芩苷2.049~2.663 mg/g、黄芩素0.018~0.032 mg/g、汉黄芩素0.021~0.035 mg/g、盐酸小檗碱0.809~1.32 2 mg/g、巴马汀0.365~0.652 mg/g、甘草苷1.442~1.972 mg/g、甘草酸铵0.883~1.071 mg/g;
15 批基准样品中各成分质量分数分别为葛根素8.031~10.938 mg/g、大豆苷1.048~1.789 mg/g、大豆苷元0.117~0.244 mg/g、黄芩苷4.709~7.369 mg/g、汉黄芩苷2.112~2.199 mg/g、黄芩素0.011~0.022 mg/g、汉黄芩素0.012~0.022 mg/g、盐酸小檗碱0.532%~1.154 mg/g、巴马汀0.224~0.423 mg/g、甘草苷1.188~1.954 mg/g、甘草酸铵0.876~0.992 mg/g。
2.3.14 GQD 各味药指标成分含量在饮片-水煎液-基准样品之间的传递规律 饮片-水煎液-基准样品中指标成分含量的转移率是考察传递规律的重要指标,其计算方法为饮片到水煎液转移率=m2/m1,水煎液到基准样品转移率=m3/m2,其中,m1为饮片中指标性成分的质量,m2为水煎液中指标性成分的质量,m3为冻干粉中指标性成分的质量。
根据试验样品所测数据,对各药味指标成分含量转移率结果如表6、7 所示,葛根素饮片到水煎液的转移率为17.71%~19.69%,水煎液到基准样品的转移率为84.18%~104.00%;大豆苷饮片到水煎液的转移率为10.32%~13.65%,水煎液到基准样品的转移率为86.90%~105.38%;大豆苷元饮片到水煎液的转移率为21.13%~27.65%,水煎液到基准样品的转移率为64.64%~104.09%;黄芩苷饮片到水煎液的转移率为22.46%~23.99%,水煎液到基准样品的转移率为71.58%~107.61%;汉黄芩苷饮片到水煎液的转移率为21.38%~28.16%,水煎液到基准样品的转移率为82.54%~106.78%;黄芩素饮片到水煎液的转移率为2.15%~3.81%,水煎液到基准样品的转移率为51.85%~83.33%;汉黄芩素饮片到水煎液的转移率为4.83%~9.55%,水煎液到基准样品的转移率为43.75%~73.33%;盐酸小檗碱饮片到水煎液的转移率为6.33%~8.70%,水煎液到基准样品的转移率为60.33%~99.95%;巴马汀饮片到水煎液的转移率为11.09%~15.79%,水煎液到基准样品的转移率为60.56%~76.94%;甘草苷饮片到水煎液的平均转移率为57.82%~76.53%,水煎液到基准样品的平均转移率为80.98%~103.70%;甘草酸铵饮片到水煎液的平均转移率为14.98%~18.08%,水煎液到基准样品的转移率87.96%~106.12%。
根据《征求意见稿》中相关内容,不同批次基准样品的指标性成分含量及转移率应控制在其均值的70%~130%,本实验中15 批GQD 基准样品的各指标性成分的含量及转移率均在其均值的±30%范围内,说明前期葛根、黄芩、黄连、炙甘草饮片和基准样品的制备工艺相对稳定可行。
2.4 GQD 基准样品制备过程中干膏率传递规律研究
按“2.1”项下的制备方法制备GQD 全方、葛根、黄芩、黄连、甘草各单味的水煎液,精密吸取50 mL 于100 mL 恒定质量后的蒸发皿中,水浴加热成稠膏,真空干燥48 h 以上至恒定质量,即得各饮片及15 批GQD 基准样品的干膏粉。计算15 批基准样品的实际干膏率及其对应饮片的干膏率,计算公式为饮片单煎干膏率=m1/M1,全方水煎液干膏率=m2/M2,其中m1表示饮片干膏粉质量,M1表示饮片质量,m2表示GQD 基准样品干膏粉质量,M2表示全方饮片质量。
为了解饮片-基准样品传递过程中干膏率的变化,根据单味药剂量在全方中的占比以及各单味药饮片的干膏率,计算全方中各单味药折算加和后的理论干膏率,计算公式为全方理论干膏率=∑单味药干膏率×(单味药生药量/全方生药量),对比理论干膏率于实际出膏率,计算转移率,结果见表8,具体计算公式为转移率=实际干膏率/理论干膏率。由表8 可知,15 批全方基准样品的理论干膏率为29.71%~33.31%,实际干膏率在24.63%~26.91%,干膏率的传递率均值为81.35%,转移率为75.08%~87.48%,均未出现离散数据(平均值的±10%以外),表示不同批次基准样品间干膏率较为稳定。

表8 15 批GQD 单味药饮片、基准样品的干膏率及转移率结果Table 8 Results of dry extract rate and transfer rate of each decoction pieces and benchmark samples of GQD
3 讨论
3.1 指标性成分的确定
经典名方基准样品作为后续复方制剂生产及质量控制的参照,在研究过程中具有核心地位,因此,其指标成分的选择应具有代表性,并能全面反映复方质量[25]。结合GQD 质量标志物以及相关文献报道[26-27],本研究采用化学指纹图谱与多指标成分在饮片-水煎液-基准样品传递相结合进一步确定该方与药效相关联的关键成分。葛根中具有较强药理活性且含量最丰富的成分为异黄酮类,研究表明,异黄酮类中主要的活性成分为葛根素、大豆苷、大豆苷元等,这些成分在抗氧化、降血糖、解热、抗炎、调节免疫等方面具有较好的药理作用[28]。黄芩中主要含有黄酮及其苷类化合物,其代表性活性成分为黄芩苷、黄芩素、汉黄芩苷、汉黄芩素,这些成分具有多方面的药理活性,包括抗炎、抗菌、抗肿瘤、抗氧化、降血压、提高免疫力等[29]。黄连的主要活性成分为生物碱类成分,本研究参照《中国药典》[17]收载的葛根芩连片剂、丸剂2 种剂型中明确规定的黄连的定量指标盐酸小檗碱外,还选择了巴马汀作为指标成分进一步研究。甘草的指标成分的选择依据《中国药典》甘草饮片项下的定量指标甘草苷、甘草酸铵。因此,选择测定葛根素、大豆苷、大豆苷元、黄芩苷、黄芩素、汉黄芩苷、汉黄芩素、盐酸小檗碱、巴马汀、甘草苷、甘草酸铵11 种与葛根芩连汤药效相关且较稳定的指标成分的含量,能进一步提升对GQD 内在质量的控制。
3.2 供试品溶液制备方法考察
在供试品制备过程中,本研究对提取溶剂(50%甲醇、70%甲醇,甲醇、50%乙醇、70%乙醇、乙腈、纯水)、提取方法(超声、室温)、提取时间(20、25、30 min)和溶剂用量(15、20、25、30 mL)等影响因素进行了考察,从提取化学成分全面性以及方法稳定性等方面考虑,确定GQD 基准样品及单味饮片供试品溶液最佳制备方法为以70%甲醇为提取溶剂,溶剂用量为25 mL,超声提取30 min。
3.3 色谱条件分析
3.3.1 GQD 基准样品指纹图谱的色谱条件考察通过查阅相关文献数据[17],分别以乙腈-0.5%磷酸水溶液、乙腈-0.05%磷酸(含0.01 mol/L 磷酸二氢钾)水溶液、乙腈-0.05%磷酸二氢钾+0.05%三乙胺溶液(磷酸调pH 至3.0)、乙腈-0.02 mol/L 醋酸铵+0.03%三乙胺溶液(用冰醋酸调pH 至4.3)共4 种不同流动相体系和比例下色谱峰的分离情况,结果显示以流动相乙腈-0.02 mol/L 醋酸铵+0.03%三乙胺溶液(用冰醋酸调pH 至4.3)进行梯度洗脱时,基线平稳,色谱峰分离度良好;参考《中国药典》以及相关文献报道[10,16],选择230、250、280、346 nm 4 个波长作为测试波长,显示在280 nm 时各色谱峰响应较好,杂质干扰少;另外,考察了柱温(25、30、35 ℃)对色谱峰分离的影响,结果发现在30 ℃条件下,各色谱峰分离度较好。
3.3.2 GQD 4 味药指标成分含量测定的色谱条件考察 由于GQD 中化学成分复杂多样,主要为黄酮类、生物碱类和皂苷类成分,通过全波长扫描发现黄芩、葛根以及甘草的指标成分在230~280 nm均有较强吸收,而黄连的生物碱类成分在330~360 nm 处有较强吸收,因此,采用同一波长不能同时实现不同成分的最大吸收。该研究曾采用指纹图谱色谱条件,通过切换波长来实现对多成分含量同时测定,但实际考察过程中发现切换波长对基线、峰形有一定的影响,基线较差对含量测定的精度有一定影响。故本研究在参考《中国药典》的基础上,分别建立了方中4 味药的含量测定方法。
此外,通过查阅文献发现,HPLC 法测定黄酮类化合物时,流动相多为乙睛-水系统或甲醇-水系统[30-31],并通常在水相中加入0.1%冰醋酸、磷酸或甲酸,以抑制黄酮中酚羟基的电离,改善拖尾,并提高分离度[32]。因此,在参考《中国药典》的基础上,结合预试验考察,进一步优化了葛根、黄芩、黄连、甘草的色谱条件,最终建立了4 味药的含量测定方法。
3.4 15 批GQD 基准样品的指纹图谱分析
15 批GQD 基准样品的指纹图谱相似度良好(>0.99),匹配结果显示出有34 个共有峰,并指认出13 个特征峰。方中的葛根、黄芩、黄连、炙甘草在《中国药典》2020 年版单味药项下规定的指标性成分均指认出,表明所建立的指纹图谱可以基本满足表征每味药特征峰的要求。然而,由于单味饮片煎液及其基准样品的供试品溶液成分更为复杂,使得黄连的指标性成分表小檗碱与黄连碱在该色谱条件下分离度并不理想。同时,该色谱条件下的30~45 min 并未检测到主成分,仍可进一步优化。对于上述的不足之处,本课题组今后将进一步深入研究,力求方中各味药的主成分出峰时间更为合理,分离度更佳。
3.5 15 批GQD 基准样品量质传递规律分析
11 种指标性成分在饮片至水煎液的传递过程中,成分的转移率皆在规定范围(均值的70%~130%)内,说明该煎煮过程较稳定可控,工艺可行。其中,甘草酸铵、盐酸小檗碱、巴马汀转移率较低,通过对比研究发现,黄连、甘草饮片单煎液皆较为澄清透明,而复方汤液明显有大量絮状物生成,通过查阅文献推测这些成分在复方汤液煎煮过程中发生酸碱络合反应,以絮状物的形式被滤过除去部分,造成了成分损失[33-34]。
此外,黄芩素、汉黄芩素转移率也较低,通过查阅文献发现,黄芩素与汉黄芩素本身不稳定,易被氧化成醌类,且黄芩素和汉黄芩素是由黄芩中所含的一种酶水解黄芩苷和汉黄芩苷而成,此酶在冷水中活性较高,热水中活性较低[35],因此,其在煎煮过程中导致成分含量的下降。在水煎液-基准样品的过程中,部分批次的转移率有些许升高,大于100%,推测在水溶液和冻干粉两者不同的物理状态下,导致虽在相同的溶剂和提取方法下检测到的含量仍有些许差异,但11 种指标成分在水煎液-基准样品的转移率皆在规定范围(均值的70%~130%)内,说明该基准样品的制备工艺稳定可行,成分具有良好传递性。该研究为后续GQD 基准样品的质量控制提供了参考。
利益冲突所有作者均声明不存在利益冲突
