动脉粥样硬化中的表观遗传机制及治疗作用*
2024-02-29童晓岚宋佳欣姜继宗
童晓岚 ,宋佳欣 ,姜继宗†
①上海大学 医学院,上海器官修复工程技术研究中心,上海 200444;②上海大学 生命科学学院,上海 200444
动脉粥样硬化(atherosclerosis, As)是引发多种心血管疾病的重要因素,而As诱导的心血管疾病是造成死亡的主要原因之一[1]。As的遗传易感性被认为是一个风险因素,但全基因组关联分析结果显示只有10.6%的病例与遗传基因相关,这表明其他因素也对As的发生发展产生影响[2]。随着对心血管疾病在分子水平研究上的不断深入,更多As的新机制被揭示,如内皮细胞功能障碍[3]、脂质氧化和炎症[4]、剪切应力学说等[5]。其中,DNA甲基化、组蛋白修饰、非编码RNA (ncRNA)调控等表观遗传机制可能参与了As的发病机制[6]。表观遗传调控是指不改变DNA基本序列,通过DNA共价修饰和基因转录后的修饰调节基因表达。表观遗传机制已被证明可以改变人类基因组与环境之间的相互作用。阐明表观遗传与As之间的关系,将有助于对As疾病的深度理解,为临床开发治疗As的新靶点提供参考。本文就表观遗传在As中的调控机制与治疗作用进行综述,旨在为As的临床治疗提供新思路和新途径。
1 动脉粥样硬化
As是一种慢性炎症疾病[7],通常伴有复杂的病理变化,如脂质代谢异常、炎症细胞因子水平升高和内皮功能障碍等不良反应。As的第一阶段是机体在外界环境因素刺激下产生氧化应激,导致血浆低密度脂蛋白(low density lipoprotein, LDL)被氧化形成氧化低密度脂蛋白(oxidized low density lipoprotein, ox-LDL)。随后,活化的巨噬细胞吞噬ox-LDL后产生泡沫细胞,引起机体病变[8]。巨噬细胞活化后释放炎症因子,促进炎症的发生[9]。第二阶段,在炎症状态下,血管平滑肌细胞(vascular smooth muscle cell, VSMC)由外膜向内膜转移,形成包裹As斑块的稳定纤维帽,导致动脉阻塞。持续性的炎症会促进As斑块形成,该斑块由死亡的平滑肌细胞、巨噬细胞和泡沫细胞组成[10]。第三阶段,基质金属蛋白酶(matrix metalloproteinase, MMP)被激活,会破坏斑块稳定性,使斑块破裂,最终导致心肌梗死或脑梗死[11]等心脑血管疾病。
2 表观遗传调控
表观遗传调控是可逆的,通过DNA甲基化、组蛋白修饰、ncRNA调控、染色质结构重塑等机制来调控基因的表达可影响疾病进展。表观遗传调控的特点是影响基因表达而不改变DNA序列。表观遗传调控与心血管[12]、神经系统[13]、代谢异常[14]等疾病的发生发展密切相关。
2.1 DNA甲基化
DNA甲基化是最稳定、研究最为广泛的一种表观遗传修饰,在基因表达调控中发挥重要作用。DNA甲基化作为一种常见的共价修饰,一般出现在CpG岛上。CpG岛是DNA短区域内密集且数量众多的CpG位点,主要位于启动子区域,通常为非甲基化状态[15]。DNA甲基化主要包括两类,分别是由DNA甲基转移酶(DNMT)1/2介导的维持甲基化和DNMT3A/3B/3L介导的从头甲基化组成。在正常个体的基因组中,基因启动子区的CpG岛一般表现为低甲基化,而非启动子区域中的CpG岛呈现高甲基化。DNA甲基化在维持细胞正常生理功能、促进胚胎发育等方面具有重要意义,一旦DNA甲基化发生紊乱,就会导致许多疾病[16]。
2.2 组蛋白修饰
组蛋白上的修饰统称为组蛋白翻译后修饰,主要在组蛋白核小体N-末端尾部进行,包括乙酰化、甲基化、泛素化、丝氨酸的磷酸化和精氨酸甲基化等[17]。与DNA甲基化相反,大多数组蛋白翻译后修饰是高度动态的过程[18],在DNA修复、选择性剪接、基因转录调控及染色体浓缩方面发挥重要作用。这些修饰依赖表观遗传因子组蛋白乙酰转移酶(histone acetyltransferases, HATs)、组蛋白脱乙酰酶(histone deacetylases, HDACs)、组蛋白甲基转移酶(histone methyltransferases, HMTs)等,直接或间接地参与调节血管、免疫和其他组织特异性基因的表达。组蛋白乙酰化能减少赖氨酸残基上的负电荷,减弱组蛋白与DNA结合,从而暴露出潜在DNA,促进基因的转录[19]。组蛋白的磷酸化与乙酰化类似,也是高度动态的过程。组蛋白甲基化主要出现在精氨酸和赖氨酸的侧链上,不同于乙酰化和磷酸化的高度动态性,组蛋白甲基化对组蛋白的电荷没有影响,是一种稳定的静态修饰。
2.3 ncRNA调控
ncRNA根据其大小和形状可分为不同的类别,目前的研究主要集中在微小RNA(microRNA,miRNA)、长链非编码RNA(long non-coding RNA,lncRNA)和环状RNA(circular RNA, circRNA)。其中,miRNA是参与基因表达转录后调控的约22 nt长的非编码序列,也是研究最为广泛的ncRNA。众多研究表明,ncRNA在调控细胞增殖、分化、凋亡等多个生物学过程中发挥着重要作用[20],同时也参与许多疾病的调控[21],如心血管系统疾病、癌症[22]和肿瘤等[23-24]。
3 表观遗传调控动脉粥样硬化
表观遗传对环境刺激高度敏感,其中DNA甲基化、组蛋白修饰和非编码RNA作为表观遗传的主要机制在环境因素影响下易受到影响。通过表观遗传调控因子DNMTs、HATs、HDACs等的调控,引起机体内参与As发生发展的关键细胞(内皮细胞、平滑肌细胞和巨噬细胞)病理性变化,促进As进展。表观遗传对As的调控机制总结如图1所示。
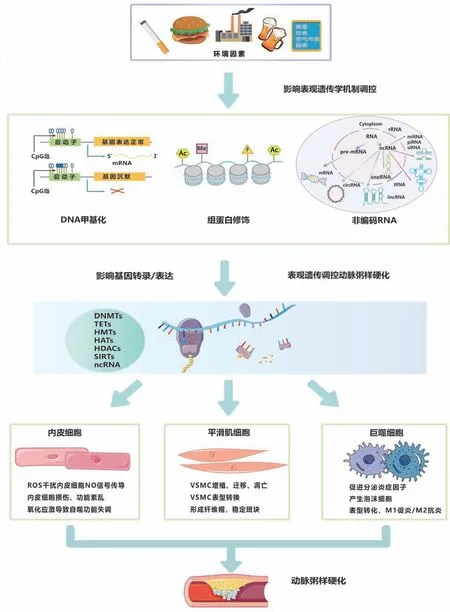
图1 表观遗传调控动脉粥样硬化示意图
3.1 DNA甲基化介导的动脉粥样硬化
DNA甲基化紊乱与As的发生发展密切相关。DNA甲基化紊乱可导致内皮细胞功能障碍、巨噬细胞介导的炎症发生、VSMC异常增殖、斑块破裂和血栓形成,从而加重As[25]。由单核巨噬细胞浸润而发生的慢性炎症是As发展的重要标志。垂死的巨噬细胞释放其脂质内容物和组织因子,形成促血栓性坏死核心,这是导致斑块不稳定的关键成分[26]。在As斑块中,巨噬细胞分泌多种促炎因子,从而形成炎症环境。单核细胞在受到斑块微环境的刺激后,可分为两类亚群,分别是具有促炎(杀伤)活性的经典M1表型和具有抗炎(修复)活性的替代M2表型。As的发展通常与巨噬细胞表型的动态变化有关,研究表明DNA甲基化参与了巨噬细胞的表型调控。Krüppel样因子4 (Krüpple-like factor 4, KLF4)和Krüppel样因子2 (Krüpple-like factor 2, KLF2)属于锌指调节转录因子,能够调节炎症和维持内皮细胞稳态,使血管内皮细胞具有抗炎及抗血栓作用。KLF4在As发生过程中扮演重要的角色,参与泡沫细胞形成、VSMC表型转化、巨噬细胞极化、内皮细胞炎症和淋巴细胞分化[27]等过程。若KLF4的部分功能出现障碍,将促进血管细胞黏附分子1 (vascular cell adhesion molecule 1, Vcam1)的表达及炎症细胞蓄积[28],加剧疾病进展。研究表明血流紊乱诱导KLF4启动子上的DNMT3A富集,导致KLF4启动子内CpG岛的DNA甲基化,抑制KLF4转录,减弱了其对As炎症的抑制作用。阻断KLF4的上游DNA甲基化,可以改善血流紊乱诱导的促炎和促血栓效应[29-32],起到抵抗As的作用。
同型半胱氨酸(homocysteine, Hcy)水平增高是As的独立危险因素。Hcy能够通过炎症反应和DNA甲基化紊乱来促进As发生[33]。已有研究表明雌激素对As的发展具有抑制作用,通过抑制VSMC增殖可减缓As进展[34]。雌激素对血管保护是依赖雌激素受体发挥作用的。雌激素通过激活雌激素受体α(estrogen receptor α, ERα)介导的自噬来减轻内皮细胞焦亡,从而预防As[35]。Hcy诱导平滑肌细胞(smooth muscle cell, SMC)雌激素受体基因启动子区域的从头甲基化,对平滑肌细胞增殖具有促进作用。SMC的增殖与As斑块的增厚和坏死核心的形成有关,雌激素受体基因高甲基化会促进As形成[36]。另有研究表明Hcy能诱导SMAD7启动子高度甲基化,从而促进血管炎症反应。与正常动脉壁相比,As斑块中的SMAD7表达降低,SMAD7启动子在As患者中表现为高度甲基化,这与Hcy水平和颈动脉斑块评分呈正相关。因此,SMAD7甲基化可能是As的新生物标志物和治疗靶点[37]。随着Hcy水平的升高,可激活NF-κB信号并抑制DNMT1活性,使血清单核细胞趋化蛋白1 (monocyte chemotaxis protein-1, MCP-1)启动子DNA低甲基化,也会诱导As斑块的形成[38]。ox-LDL在巨噬细胞中可激活DNMT1。DNMT1水平的升高诱导过氧化物酶体增殖物激活受体γ (peroxisome proliferatoractivated receptor γ, PPARγ)启动子发生高甲基化,促进As发生发展[39]。组织因子途径抑制物-2(tissue factor pathway inhibitor-2, TFPI-2)是在As斑块中表达的一种Kunitz型丝氨酸蛋白酶抑制剂,参与抑制MMP的活性[40]。在As中,MMP可以降解动脉细胞外基质的胶原蛋白,使斑块不稳定。单核细胞/巨噬细胞过度产生MMP可促进As斑块破裂并诱导心肌梗死发生[41]。MMP-9是导致斑块破裂的重要MMP之一,也是急性心肌梗死的重要标志物[42]。研究表明,在As斑块中TFPI-2基因发生甲基化,使得TFPI-2基因表达降低,进一步促进了细胞外基质的降解和As进展[43]。
综上所述,异常的DNA甲基化可能会影响As相关基因的表达,这些基因有望成为临床上干预As治疗的新靶点[17]。
3.2 组蛋白修饰介导的动脉粥样硬化
在As中,组蛋白乙酰化和甲基化发生了显著变化,从而影响染色质结构和基因转录。研究发现,As病变中SMC和巨噬细胞的组蛋白H3上的K9和K27位置(H3K9和H3K27)显示出更高的乙酰化水平,导致血管壁内的病理变化,促进As的发生或进展。在SMC和淋巴细胞中,H3K9和H3K27的甲基化显著降低[44],进而激活SMC。一方面,SMC通过产生新的胶原纤维来稳定血管壁;另一方面,这些细胞也过度表达各种MMP和炎症因子,促进斑块不稳定,导致斑块破裂[45]。此外,血管紧张素Ⅱ (angiotensin II, Ang II)通过组蛋白乙酰转移酶共激活剂p300和SRC-1,增加了白细胞介素-6(IL-6)启动子上的组蛋白H3赖氨酸乙酰化,促进炎症因子IL-6的释放[46],加剧As的进展。
Nox5是一种新型的NADPH氧化酶,可产生超氧化物,产生氧化应激促进As发生。在炎症条件下,p300和HAT1的水平增加,共同促进了Nox5基因启动子的乙酰化,这表明组蛋白乙酰化参与As发展过程[47]。通过调控表观遗传的途径,控制Nox5的表达可能是治疗As的新靶点,而不是直接清除ROS。HDAC抑制剂TSA (trichostain A)通过诱导p21表达抑制VSMC的增殖[48]。在高同型半胱氨酸血症相关的As中,HDAC的增高可使细胞内的乙酰化水平下降,并促使总胆固醇、游离胆固醇、甘油三酯在泡沫细胞中累积[49],导致脂质代谢发生异常。有研究表明MMP基因增加与H3K9乙酰化成正比[50],在As斑块中MMP的过表达可导致斑块不稳定,从而诱发斑块破裂[48,51]。SIRT1是组蛋白脱乙酰酶HDAC家族的成员,通过抑制巨噬细胞LOX-1基因表达,减少巨噬细胞对ox-LDL的吞噬,从而减少内膜下脂质沉积,延缓As进展[52]。KLF2是内皮抗炎和抗As的关键介质,HDAC5通过与KLF2直接结合抑制KLF2的转录激活,并且下调KLF2依赖性的内皮细胞一氧化氮合酶(endothelial nitric oxide synthase, eNOS)表达,导致内皮细胞功能发生障碍,诱导As的发生[53]。
综上,组蛋白修饰与As发展密切相关,相应乙酰化转移酶表达参与As的调控过程,可能作为预防心血管相关疾病的潜在靶点。
3.3 非编码RNA介导的动脉粥样硬化
ncRNA可以通过破坏稳定的靶标mRNA翻译来调节As的进程。例如:miR-195-3p通过靶向抑制炎症因子IL-31表达,来减轻炎症和As斑块的形成[54];miR-19b促进炎性细胞因子的分泌和巨噬细胞侵入内皮层,促进As的进展[55];miR-181b通过上调巨噬细胞TIMP-3表达,来减少As斑块的形成[56];miR-126能减少炎症细胞因子的释放,从而影响As的进展[57]。Hcy促进VSMC增殖是通过miR-143介导的,这对As的发病机制和进展至关重要[58];miR-221通过抑制NCoR启动子区域中DNMT3B介导的DNA甲基化,来抑制ox-LDL诱导的炎症反应[59];lncRBA GSA5可以通过miR-221的海绵机制来触发炎症反应和MMP表达,促进纤维帽降解并加重As斑块的不稳定性[60];circ_Lrp6阻碍了miR-145介导的VSMC迁移、增殖和分化的调节,起到抑制As发生发展的作用[61];lncRNA-FA2H-2的敲低和MLKL的过表达均能显著加重ox-LDL诱导的炎症反应。lncRNA-FA2H-2-MLKL通路对于调节自噬和炎症至关重要,所以lncRNA-FA2H-2和MLKL可以作为改善As相关疾病的潜在治疗靶点[62]。ncRNA在As中的调控总结如表1所示。

表1 ncRNA在动脉粥样硬化中的调控
4 表观遗传与动脉粥样硬化治疗
4.1 针对DNA甲基化的治疗策略
CpG岛甲基化后可直接干扰转录因子与DNA调控区域的结合,从而影响基因转录[63]。肝X受体α(liver X receptor α, LXRα)、PPARγ是影响巨噬细胞炎症和胆固醇稳定性的重要转录因子,在As的发展过程中起关键作用。LXRα和PPAR-γ富含CpG位点,5-氮胞苷(5-aza-2′-deoxycytidine, 5-aza-dC)作为DNA甲基转移酶抑制剂能够对LXRα和PPARγ1启动子进行去甲基化并下调参与炎症基因和趋化性因子的表达,这减弱了巨噬细胞迁移和对内皮细胞的黏附,减少巨噬细胞浸润到As斑块中。通过5-aza-dC抑制DNA甲基化可降低As斑块中的巨噬细胞内质网应激和凋亡,有助于改善As[64]。白细胞介素信号传导可促As发生[65],细胞因子IL-6在各种疾病的炎症信号通路中发挥了促炎和抗炎双重特性[66]。有研究发现As患者中的IL-6基因表达水平显著高于对照组[67],IL6启动子中的上游CpG岛呈现出低甲基化水平。使用天然的IL-6信号转导抑制物可溶性糖蛋白(soluble glycoprotein 130, Sgp130)和免疫球蛋白G1 (IgG1-Fc)的融合蛋白 (Sgp130Fc)治疗可显著改善高胆固醇血症患者的As程度,说明阻断IL-6表达可作为治疗As的新策略。此外,Sgp130Fc对IL-6转导信号的抑制作用是特异性的,不抑制其他IL-6细胞因子家族成员的活性[68]。DNMT3B介导的CREG基因高甲基化是导致内皮功能出现异常和As发展的新机制。ox-LDL通过上调DNMT3B表达水平来降低人脐静脉血管内皮细胞(human umbilical vein endothelial cells, HUVECs)中CREG的表达,因此,可通过下调DNMT3B阻断CREG甲基化来治疗As[69]。有研究发现叶酸可以通过减少ROS诱导的氧化损伤和细胞凋亡,抑制ox-LDL诱导的内皮功能障碍。这与DNMT活性增加和血管过氧化物酶1(vascular peroxidase 1, VPO1)启动子的DNA甲基化改变以及VPO1表达的变化有关。该研究表明叶酸的缺乏可导致内皮功能障碍,通过使用叶酸作为营养补充剂可预防As[70]。
4.2 针对组蛋白修饰的治疗策略
血管紧张素转化酶2(angiotensin converting enzyme 2, ACE2)过表达可减轻As,ACE2的缺乏会恶化ApoE敲除小鼠的As[71]。在接受高胆固醇饮食和阿托伐他汀治疗的新西兰白兔的心脏组织中,与对照高胆固醇饮食相比,治疗组ACE2的mRNA水平升高。如前所述,Hcy不仅会增加细胞中异常的DNA甲基化,而且还通过H3K9上的乙酰化抑制HDAC1的表达,提高NMDAR1、DNMT1和MMP-9的水平。该研究中阿托伐他汀通过改变组蛋白修饰来影响ACE2表达[72],表明调控组蛋白修饰可用于治疗As。TGF-β是一种抗As的细胞因子,可抑制免疫反应并稳定As[73]。在人类中,HDAC3是唯一在As病变中上调的HDAC。HDAC3的缺失可激活TGF-β表达并提高斑块的稳定性。HDAC3也对于调节巨噬细胞的纤维化表型至关重要。HDAC3的缺失使巨噬细胞转变为抗炎表型并减少脂质积累,靶向巨噬细胞表观基因组可以改善As[74]。细胞外超氧化物歧化酶(EC-SOD)是SOD同工酶之一,通过加速超氧化物的歧化反应在预防心血管疾病中起重要作用。从蜂王浆中分离的化合物4-HPO-DAEE通过单核细胞THP-1细胞中的组蛋白乙酰化增加EC-SOD的表达,该化合物具有作为抗As的候选药物或先导化合物的潜力[75]。组织纤溶酶原激活剂(tissue-type plasminogen activator, t-PA)是调节纤维蛋白溶解的关键酶,在As斑块破裂或局部血栓形成初始时,t-PA的快速释放能防止血栓形成。研究表明HDAC抑制剂丙戊酸能增强As患者的t-PA释放能力,降低血栓发生[76]。姜黄素具有强大的HDAC抑制活性,通过抑制VSMC的生长和增殖防止不良血管重塑发生[77],起到抗As的作用。研究表明姜黄素通过激活脱乙酰化酶SIRT1和p300/CBP表达,防止高葡萄糖诱导的内皮细胞黏附和促血管生成活化的作用,从而延缓As进展[78]。白藜芦醇是一种非黄酮类多酚植物抗毒素。与姜黄素一样,白藜芦醇通过影响许多分子靶标的活性或表达,对血管和心脏功能表现出多效性的保护作用[79]。白藜芦醇的抗炎特性主要是通过下调SIRT1介导的NF-κB释放。NF-κB是一种关键的炎症转录因子,白藜芦醇通过激活SIRT-1,使NF-κB-p65和H3K9脱乙酰化,抑制了相关的氧化应激[80],起到抗As的作用。此外,白藜芦醇衍生物TMS可以在体外和体内减轻巨噬细胞衍生的泡沫细胞中的胆固醇积累,防止As病变[81]。
4.3 针对非编码RNA调控的治疗策略
ncRNA是缺乏编码肽或蛋白质能力的RNA,可作为其他基因表达的调节剂,参与蛋白质的产生[82]。通过miR-33寡核苷酸拮抗剂治疗可以提高高密度脂蛋白(high-density lipoprotein, HDL)水平,促进反向胆固醇转运,改善As[83]。PCSK9与LDL受体结合可以使LDL发生降解,从而降低As发病率。通过给受试者静脉注射ALN-PC(一种抑制PCSK9合成的siRNA)抑制PCSK9合成,在胆固醇升高的健康志愿者中耐受性良好,同时显著降低血浆PCSK9和LDL水平。这表明通过RNA干扰抑制PCSK9合成,可降低健康个体中的LDL胆固醇浓度,从而预防As[84]。另一项研究表明通过静脉注射SMC靶向的miR-145慢病毒可以使ApoE-/-小鼠中As斑块显著减少,并增加斑块的稳定性[85]。研究证明miR-92a可以直接与KLF2和KLF4的3′UTR结合,部分逆转内皮细胞产生的炎症[86]。临床上开发了一种靶向miR-92a-3p的锁核酸MRG-110,用于临床试验。I期临床试验证明单次给药MRG-110有效地降低了人类外周血中的miR-92a水平,对心血管疾病和伤口愈合具有治疗作用[87]。姜黄素通过上调miR-126来维持内皮细胞功能并抑制血管生成,从而发挥了有效的抗As作用[88]。这些研究结果表明,基于调控ncRNA的方法在治疗As方面具有巨大潜力。
5 小结与展望
As是一个复杂的动态过程,涉及一系列连续的分子和细胞活化,其特征是血管脂质积累、免疫系统激活并伴随炎症、氧化应激导致内皮细胞功能障碍、平滑肌细胞增殖和巨噬细胞活化产生泡沫细胞,最终形成As斑块[89-90]。临床上As治疗方法存在局限性,As患者术后的生活质量并未得到明显提升,需要进一步探索治疗As的新方法。基于表观遗传的治疗是一个新的研究领域,在As的治疗中,DNA甲基化、组蛋白修饰和ncRNA调控参与了该疾病的发病机制。随着As的表观遗传机制逐渐被揭示,这给开发针对染色质结构的小分子表观遗传药物,如HDAC抑制剂、Sirtuin激活化合物、DNA甲基化抑制剂和组蛋白甲基化抑制剂,来对抗As提供了新的启示。表观遗传作为基因调控的重要机制,为As的发病机制、早期诊断、新型治疗等提供了新的思路,因此未来需要进行更多表观遗传调控动脉粥样硬化的机制研究,以期寻找有效的新治疗靶点,进一步降低心血管疾病发生率和死亡率[91]。
