基于图神经网络和随机森林的CircRNA-疾病预测
2024-02-28王波尹帅杜晓昕张剑飞周振宇
王波,尹帅,杜晓昕,张剑飞,周振宇
基于图神经网络和随机森林的CircRNA-疾病预测
王波,尹帅,杜晓昕,张剑飞,周振宇
(齐齐哈尔大学 计算机与控制工程学院,黑龙江 齐齐哈尔 161006)
环状RNA(CircRNA)广泛参与人类疾病的进程,其突变和失调与许多人类疾病密切相关.因此,建立一个高效准确的CircRNA与疾病之间的预测算法对于提前对疾病的发生做出预防以及发病后的治疗方案具有重要意义.提出了一种新的基于图神经网络和随机森林的算法预测CircRNA-疾病关联算法,在分层网络表示嵌入部分通过构建异构网络,根据网络图的邻近性,对网络图的节点和边缘进行分层,递归地合并原始图中的节点和边,得到若干具有相似特征的较小子网络.子网络规模随着分层的深入而递减,直至得到最小子网络后,使用node2vec网络图游走算法对其进行预处理,然后将全部节点的特征向量输入至随机森林分类器来识别潜在的CircRNA-疾病关联,从而进行预测.
CircRNA-疾病关联预测;图神经网络;node2vec;随机森林
环状RNA(Circular RNA,CircRNA)是一类通过反向剪接产生,具有闭环结构特殊的单链非编码内源性RNA分子[1].随着高通量测序技术的发展,CircRNA已经被证明在各种生物中广泛存在,并受到广泛关注[2-4].重要的是,CircRNA的表达水平通常为组织特异性和细胞型特异性[5].因此,CircRNA的错误表达可以导致异常的生理过程,并导致大多数疾病的发生和发展[6].然而,目前对疾病与CircRNA关系的研究高度依赖生物实验,耗时耗力且效果不一定好.在这种情况下,只能发现有限数量的关系.但庆幸的是,一些CircRNA-疾病相关联的数据库已经可用,如CircRNA Disease[7]和CircR2 Disease[8],其数据库的建立为后续科研工作者使用计算模型进行潜在疾病的关联预测提供了有力保障.
目前,许多计算模型被用于挖掘潜在的CircRNA-疾病关联对.但由于当前数据集的不完备和计算模型的缺陷,要获得足够准确的预测结果仍然是一个挑战.本文使用图神经网络中的分层网络图表示算法,对基因与疾病组成的异构网络中的边缘和顶点进行合并,从而提取出节点特征.然后用Node2Vec算法学习表示成嵌入向量,并通过该嵌入向量来反映整个网络图的结构,计算出顶点间存在边缘的概率,对CircRNA-疾病网络图中的各节点的关系进行预测分析,从而达到CircRNA-疾病关联预测的目的.
1 数据集和方法
1.1 数据集
1.1.1 CircRNA关联数据集 研究中使用的CircRNA-疾病关联来自于MNDR数据库,本文从MNDR数据库提取出文献中已知的2 392个CircRNA与1 015个疾病之间的3 221个关联.MNDR V3.0是一个储存疾病相关ncRNA的全面数据库,通常被当作CircRNA与疾病关联预测的基础数据库.
1.1.2 疾病关联数据集 本文的miRNA-疾病关联数据来自HMDD V3.2数据库.HMDD是一个手工收集整理的miRNA与疾病相关联的数据库,最新版本为V3.2.在此数据库中原始数据包括两种类型,一种是在疾病过程中miRNA的主观因果关系,另一种是疾病过程中miRNA的被动变化.在实验中仅选取数据库中与疾病具有因果关系的相关miRNA-疾病关联记录,提取出664个miRNA与895种疾病之间的35 547个关联.
1.1.3 miRNA关联数据集 已知的miRNA-CircRNA关联数据集可以从starBase数据库下载,starBase数据库中记录了miRNA与各种RNA之间的联系信息,并在此基础上构建了ceRNA网络.在消除冗余后只选择CircRNA-疾病数据中常见的CircRNA记录和miRNA数据中常见的miRNA记录,最终共获得641个miRNA与865个CircRNA之间的18 317个关系.
1.2 研究方法
构建CircRNA-疾病-miRNA之间相互关联异构网络,然后通过网络图的一阶邻近性和二阶邻近性,对网络图的节点和边缘进行分层,递归地合并原始图中的节点和边,得到一系列结构相似的连续较小子网络图.使用Node2vec算法对最小子网络图进行预处理,获取顶点的低维特征向量表示,并将其输入上一层更复杂网络的特征表示中.重复这个过程直到得到原始图中每个节点的低维特征向量表示,然后将全部节点的特征向量输入至随机森林分类器来识别潜在的CircRNA-疾病关联,最终得到相应的预测结果(见图1).
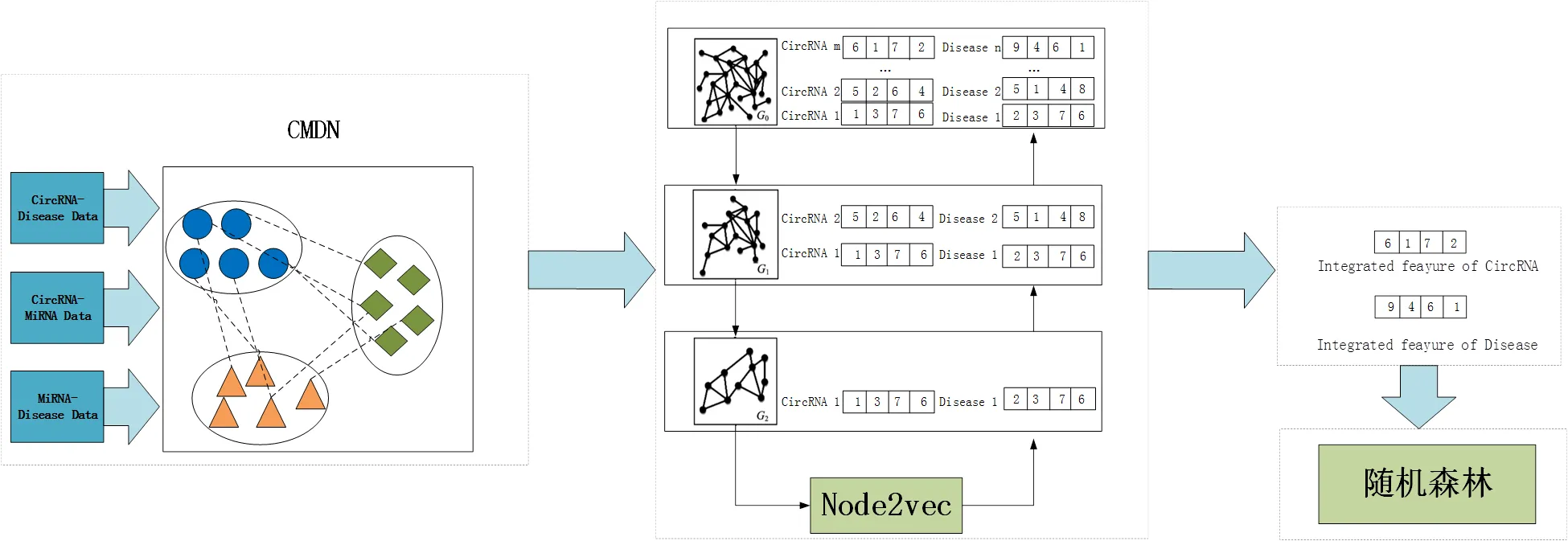
图1 算法主要流程
1.3 构建CircRNA-疾病-miRNA关联异构网络CMDN
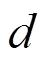

1.4 算法定义描述
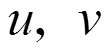
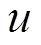
1.5 分层网络图表示学习
在网络图表示学习中普遍的任务是根据网络中顶点的邻近性,将网络中的节点通过游走类算法(如DeepWalk,Node2Vec,LINE)学习表示成嵌入向量,并通过该嵌入向量来反映整个网络图的结构,计算出顶点间存在边缘的概率,从而对网络图中的各节点的关系或链路进行预测分析[10].
但是这些网络表示学习方法一般都存在共同的问题:
(1)考虑的重点基本都聚焦在网络的局部结构关系,如节点的低阶邻近性,尽管使用了随机游走,但是也局限在特殊点附近,忽略了网络图中高阶结构特性.
(2)都是通过随机梯度下降的方法对非凸的目标函数进行优化,容易将焦点停留在局部最优点.
所以在本文中使用分层网络表示学习算法,通过递归将网络中的节点和边进行合并处理,将原始的CMDN网络进行分层处理,使其划分生成一系列分层的结构较小的网络图,然后通过现有的Node2vec算法进行不断的网络嵌入来进行特征提取.将学习到最小网络嵌入作为更大网络的初始化向量,迭代求解上一层较大网络的嵌入,不断向上循环迭代直至求解到全部的CMDN网络的嵌入,避免了随机初始化导致的局部最小值的风险.在分层的过程中主要利用网络图的图粗粒度化,图粗粒度化主要包括边缘合并和顶点合并,其中边缘合并可以保留网络中的一阶相似度,顶点合并可以保留网络中的二阶相似度.
其中网络图分层算法流程为:
1 Input:网络图CMDN=(V,E)
2 n=0

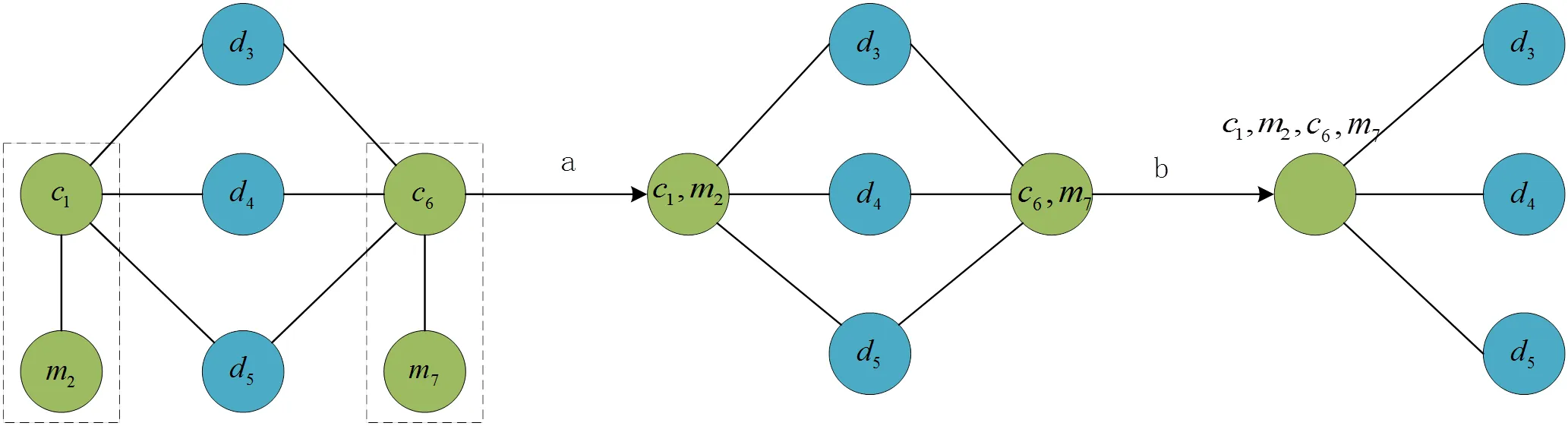
图2 网络图分层顶点合并和边缘合并算法实例
1.6 基于Node2vec的特征提取
Node2vec是一种半监督的网络特征学习方法,是在DeepWalk基础上提出的一种结合了广度优先搜索(BFS)和深度优先搜索(DFS)的有偏随机游走模型[11],两种搜索模式见图3.
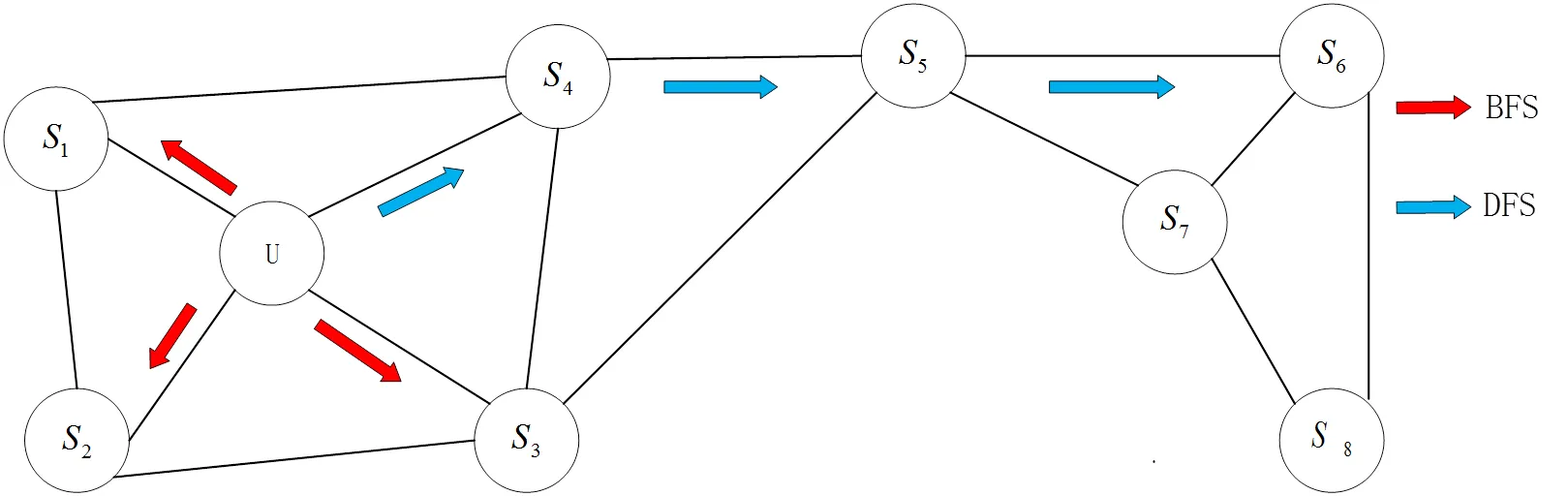
图3 BFS和DFS之间区别说明
与传统的随机游走模型不同的是,顶点序列的采样策略不同,根据实际任务的不同,会采取有偏好的调整参数来调整.不同的游走策略程度,即控制BFS和DFS的程度[12](见图4).给出Node2vec中有偏随机游走算法实现的描述:
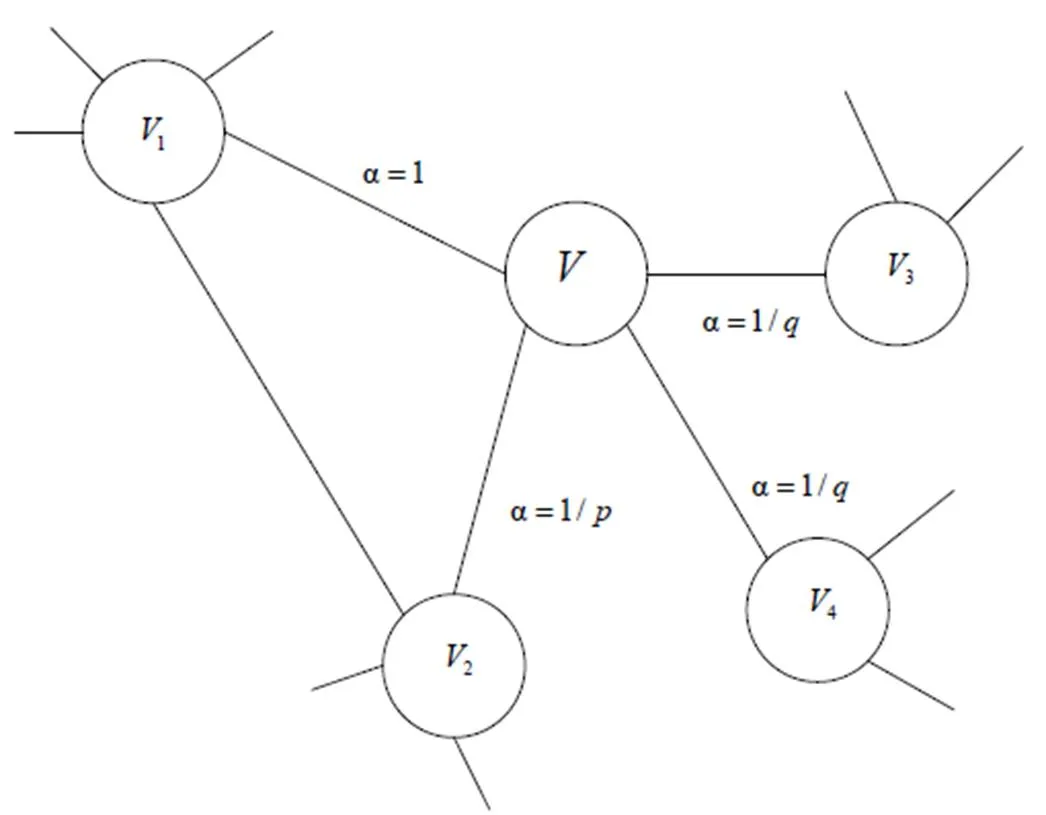
图4 有偏游走Node2vec

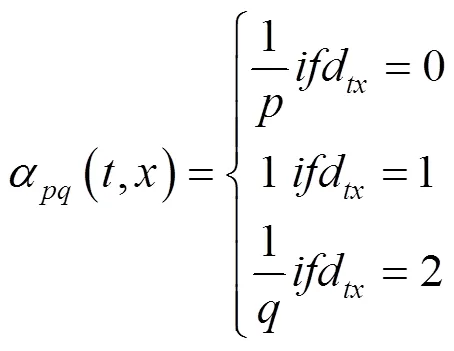
2 结果和讨论
2.1 评估指标

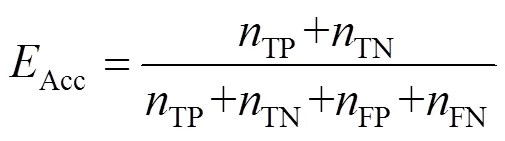
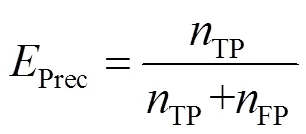
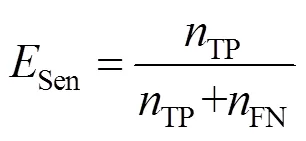
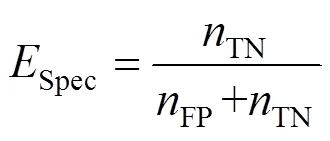
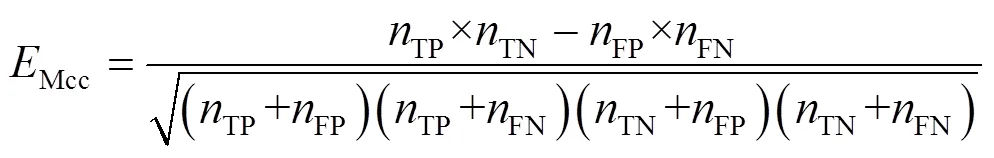
2.2 分类器的选择和参数调整
在机器学习分类器的选择过程中,分别基于不同的评估标准测试了随机森林分类器(RF)、线性回归分类器(LR)、XGBoost分类器、AdaBoost分类器(ADB)、朴素贝叶斯分类器(NB).所有分类器都是从Scikit-learn库中导入到PyCharm中实现,其内部参数均为默认值不做修改,所评估的指标均为十折交叉验证下的平均值.所有分类器的不同评估指标下的结果见表1.
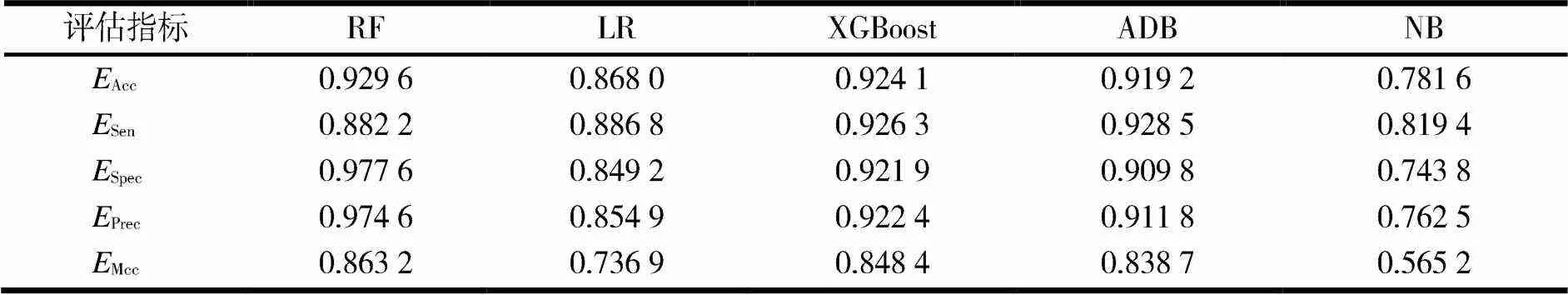
表1 分类器在不同评估指标下的结果
由表1可见,随机森林分类器(RF)在大部分评估指标下都取得了最佳的分类结果,所以在模型的机器学习分类器中采用随机森林分类器来进行分类.
2.3 模型性能评估
模型是在MNDR数据集中实现的,用于评估CircRNA-疾病潜在的关联,模型在十折交叉验证的结果见表2.
从模型在评估指标的表现来看,所有的标准差都小于2,表明模型具有良好的鲁棒性,能够很好地完成CircRNA-疾病关联预测的任务.
此外,在PyCharm中绘制了模型生成的ROC曲线(见图5),模型的ROC曲线具有很好的表现,十折交叉验证的平均AUC值为0.983 2,可以很好地进行CircRNA-疾病潜在的关联预测.
表2 模型在十折交叉验证下的产生的结果(%)
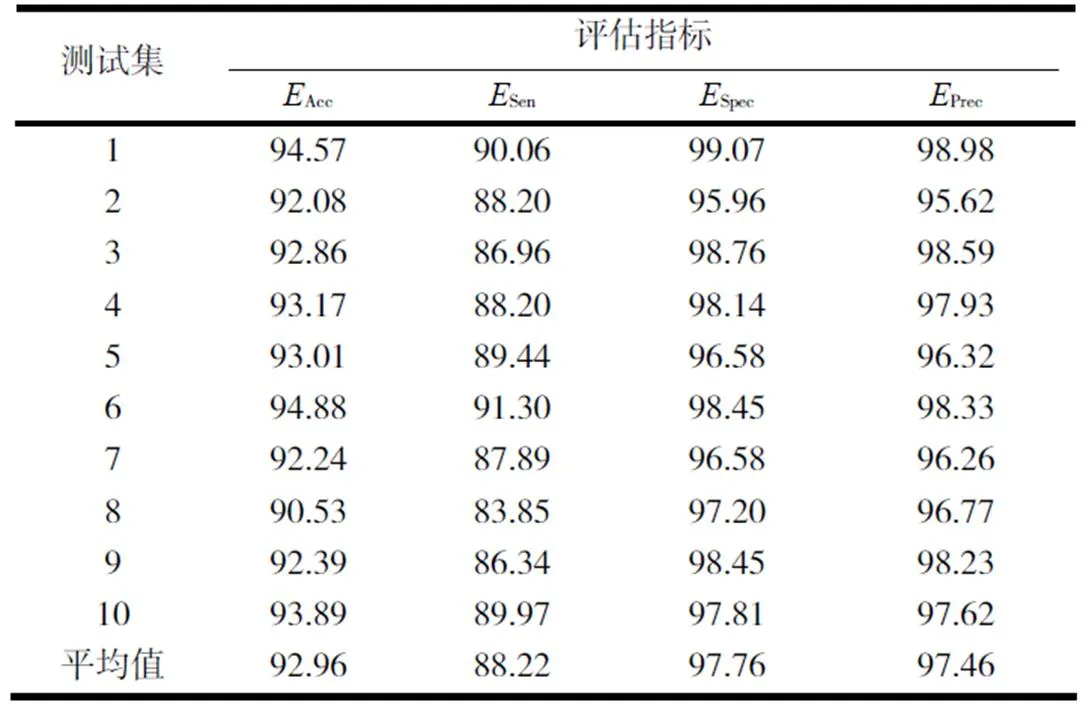

图5 模型在十折交叉验证下的ROC 曲线
对当前比较先进的几种模型在同一数据集下进行了对比,其中包括GCNCDA[13],DWNN-RLS[14],PWCDA[15],KATZHCDA[16],结果见表3.根据十折交叉验证的AUC分数来看,本文模型取得了最高的AUC分数,比对照组中表现最好的模型AUC值约提高了6%.

表3 不同模型在MNDR同一基准数据集下十折交叉验证的平均AUC分数
3 结语
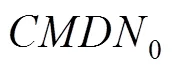
[1] ZHANG Y,ZHANG X O,CHEN T,et al.Circular Intronic Long Noncoding RNAs[J].Molecular Cell,2013,51(6):134-137.
[2] Danan M,Schwartz S,Edelheit S,et al.Transcriptome-wide discovery of circular RNAs in Archaea[J].Nucleic Acids Research,2011,40(7):3131-3142.
[3] CHEN L,HUANG C,WANG X,et al.Circular RNAs in Eukaryotic Cells[J].Current Genomics,2015,16(5):1-2.
[4] CHU Q,ZHANG X,ZHU X,et al.PlantcircBase:A Database for Plant Circular RNAs[J].Molecular Plant,2017,10 (8):1126-1128.
[5] LIANG D,WILUSZ J E.Short intronic repeat sequences facilitate circular RNA production[J].Genes & Development, 2014,28(20):2233-2247.
[6] ZHANG Z,YANG T,XIAO J.Circular RNAs:Promising Biomarkers for Human Diseases[J].EBioMedicine,2018,34: 267-274.
[7] ZHAO Z,WANG K,WU F,et al.CircRNA disease:a manually curated database of experimentally supportedCircRNA-disease associations[J].Cell Death & Disease,2018,9(5):1-2.
[8] FAN C,LEI X,FANG Z,et al.CircR2Disease:a manually curated database for experimentally supported circular RNAs associated with various diseases[J].Database,2018: 8(4):1-6.
[9] TANG J, QU M, WANG M, et al. Line: Large-scale information network embedding[C]//Proceedings of the 24th international conference on world wide web.2015:1067-1077.
[10] 高宏屹,张曦煌,王杰.生成对抗式分层网络表示学习的链路预测算法[J].计算机工程,2021,47(2):60-68,76.
[11] Grover A,Leskovec J.Node2vec:Scalable Feature Learning for Networks[J].ACM,2016.DOI:10.1145/2939672.2939754.
[12] 姚锐.采用Node2vec模型对网络特征表示方法研究[D].南京:南京大学,2018.
[13] WANG L,YOU Z H,LI Y M,et al.GCNCDA:A New Method for Predicting CircRNA-Disease Associations Based on Graph Convolutional Network Algorithm[J].Cold Spring Harbor Laboratory,2019(5):934-947.
[14] YAN C,WANG J,WU F X.DWNN-RLS:Regularized least squares method for predicting CircRNA-disease associations[J].BMC Bioinformatics,2018.DOI:10.1186/s12859-018-2522-6
[15] XIU JUAN,LEI,ZENGQIANG,et al.PWCDA:Path Weighted Method for Predicting CircRNA-Disease Associations[J]. International Journal of Molecular Sciences,2018,36:231-235.
[16] FAN C,LEI X,WU F X.Prediction of CircRNA-Disease Associations Using KATZ Model Based on Heterogeneous Networks[J]. International Journal of Biological Sciences,2018,14(14):111-114.
CircRNA-disease prediction based on graph neural networks and random forests
WANG Bo,YIN Shuai,DU Xiaoxin,ZHANG Jianfei,ZHOU Zhenyu
(School of Computer and Control Engineering,Qiqihar University,Qiqihar 161006,China)
Circular RNA(CircRNA)are widely involved in human disease processes,and their mutations and dysregulation are closely associated with many human diseases.Therefore,establishing an efficient and accurate prediction algorithm between CircRNA and diseases is important for making prevention of disease occurrence in advance as well as treatment programs after the onset of diseases.A new algorithm based on graph neural network and random forest is proposed to predict CircRNA-disease association algorithm,in the hierarchical network representation embedding part by constructing a heterogeneous network,according to the proximity of the network graph,the nodes and edges of the network graph are layered,and the nodes and edges in the original graph are merged recursively to obtain a number of smaller sub-networks with similar characteristics,and the size of the sub-networks decreases with deeper layering until the smallest sub-network is obtained.The size of the sub-networks decreases with the depth of layering until the smallest sub-network is obtained, which is preprocessed using the node2vec network graph wandering algorithm,and then the feature vectors of all the nodes are inputted into the random forest classifier to identify potential CircRNA-disease associations and thus make predictions.
CircRNA-disease association prediction;graph neural network;node2vec;random forest
TP399
A
10.3969/j.issn.1007-9831.2024.02.007
1007-9831(2024)02-0036-07
2023-07-31
2022年度黑龙江省省属高等学校基本科研业务费科研项目(145209125)
王波(1980-),男,黑龙江齐齐哈尔人,教授,博士,从事大数据分析与挖掘研究.E-mail:bowangdr@qqhru.edu.cn
