2-膦亚胺叶立德-1,4-醌类衍生物的合成
2024-02-28李福裕王继宇
汪 蓓, 胥 红, 李福裕, 王继宇
(1. 中国科学院 成都有机化学研究所,四川 成都 610041; 2. 西华大学 化学系,四川 成都 610039; 3. 中国科学院大学,北京 100049)
有机磷化合物广泛存在于自然界中,并应用于有机合成、农药、医药和材料等领域。其中,膦亚胺叶立德是重要的有机磷试剂,通常作为有机合成中间体[1]、金属配合物[2]、小分子催化剂[3]和聚合物骨架[4]等应用于有机合成、配位化学和材料科学领域。
随着对膦亚胺叶立德研究的不断深入,其合成策略也在不断丰富(图1)。 1919年,STAUDINGER等[5]直接通过有机叠氮化合物与三价膦化合物反应合成膦亚胺,即Staudinger反应,这是最早关于合成膦亚胺的文献报道,产物膦亚胺常被用作合成氨基化合物和亚胺化合物的合成中间体;2018年,MCNALLY等[6]通过芳杂环膦盐与叠氮化钠反应合成芳杂环膦亚胺,该方法可以实现区域选择性的芳杂环膦亚胺化合物的合成,并可作为Piriton、 Claritin和Gleevec等药物分子衍生化的工具,但其不足之处在于,底物范围仅应用于含N的杂芳环化合物;1954年,KIRSANOV[7]以伯胺和三价膦化合物为原料,在三乙胺和六氯乙烷的条件下合成膦亚胺类化合物,即Kirsanov反应,该方法使用伯胺化合物替代易爆炸的叠氮化物作为N源,反应条件更为安全,是另一种合成膦亚胺化合物的常见方法;2022年,赵剑楠等[8]通过光催化实现了三苯基膦和羟胺类化合物反应合成N-酰基膦亚胺类化合物,该方法反应条件温和,但底物范围相对有限。总体来看,这些方法可以实现不同类型底物的膦亚胺化,但受底物范围和底物合成难度的限制,Staudinger反应仍然是合成工作者们应用最广泛的方法。利用Staudinger反应合成膦亚胺的关键步骤为有机叠氮化合物的合成,目前已经实现以NaN3, DPPA, TMSN3和1H-咪唑-1-磺酰叠氮等试剂作为叠氮源[9-10]合成各种类型的有机叠氮化合物。
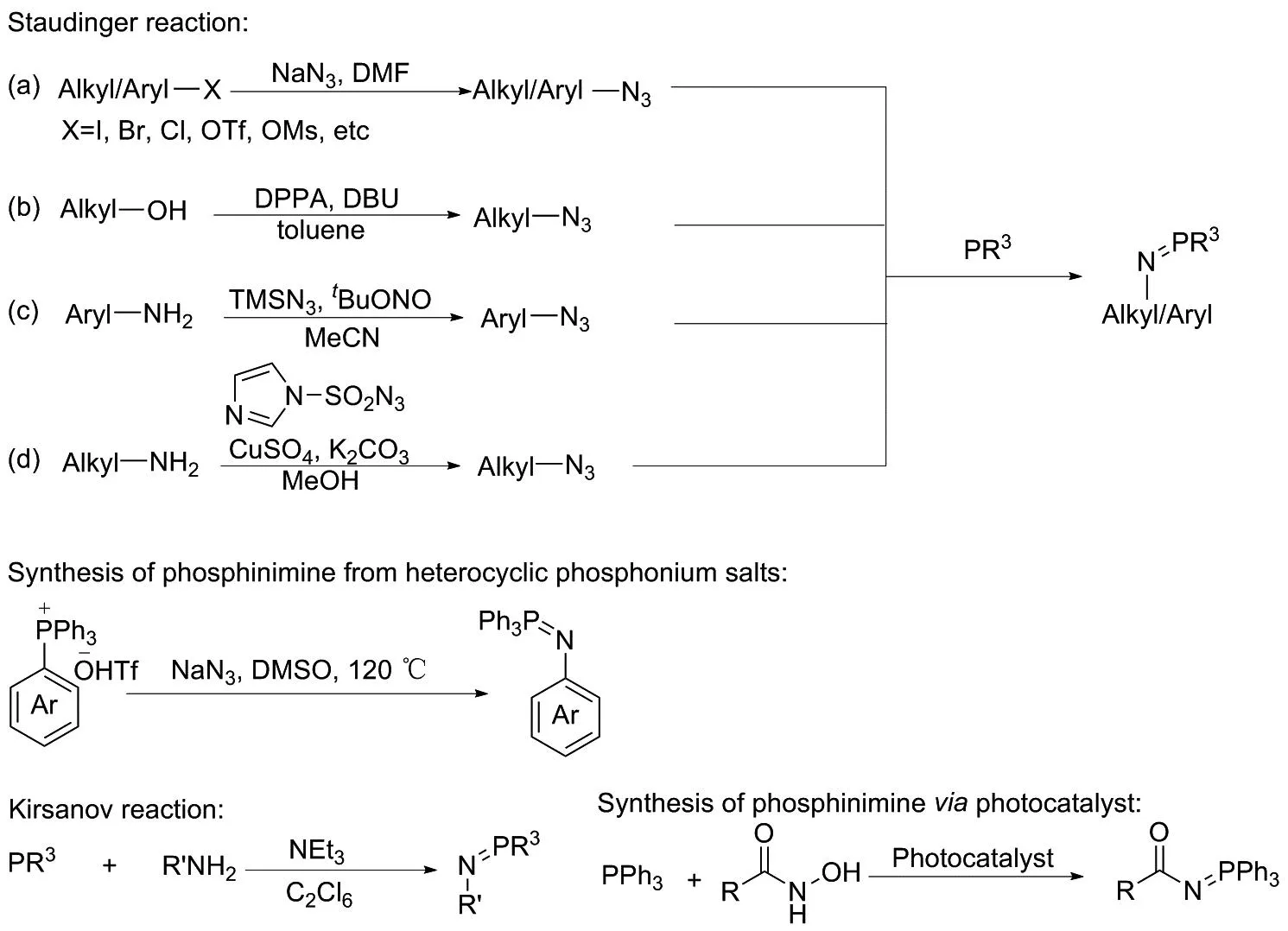
图1 膦亚胺叶立德的合成方法Figure 1 Preparation methods of phosphinimide ylide
醌类化合物是一类极其重要的有机化合物,不仅表现出抗癌、抗炎和抗疟疾等多种生理活性[11],还具有成为电极材料和荧光材料的潜能[12]。近年来,醌类化合物的官能化涵盖烷基化、芳基化、氨基化、羟基化、硫醚化和多元并环化合物的构建等多个研究方向[13-15]。但构建含膦亚胺基团的醌类化合物仅有少量文献报道[16-18]。
本文在已有研究的基础上,以1,4-醌类化合物为原料,设计开发了新的构建2-膦亚胺叶立德-1,4-醌类衍生物的合成路线(图2)。该合成路线以廉价易得的原料,通过溴代、叠氮化和Staudinger反应得到一系列2-膦亚胺叶立德-1,4-醌类衍生物,具有条件温和、操作简单和合成规模较大等优点。

图2 2-膦亚胺叶立德-1,4-醌类化合物的合成路线Figure 2 Synthesis route of 2-phosphoranylidenamino-1,4-quinones
1 实验部分
1.1 仪器与试剂
Agilent(400 MHz)型核磁共振仪(CDCl3为氘代试剂,TMS为内标); Thermo Scientific Exactive Orbitrap型高分辨质谱仪; Hanon MP430型熔点仪。
所有试剂均为国产分析纯。
1.2 合成
(1) 化合物2的合成(以2a为例)
向100 mL反应瓶中依次加入1,4-萘醌(5.00 mmol)和20 mL乙酸,将反应瓶于室温下搅拌。称取Br2(5.00 mmol, 1.00 equiv),并用5 mL乙酸稀释,将该溶液滴加入反应瓶中。反应2 h后,向反应瓶中加入无水乙酸钠(7.50 mmol, 1.50 equiv),于室温下继续搅拌30 min后,升温至110 ℃回流1.5 h。反应结束后,将反应瓶冷却至室温。向反应瓶中加入大量的水,有黄色沉淀析出,抽滤得到黄色滤饼,并用水洗涤滤饼,烘干,得到的2-溴-1,4-萘醌不经进一步纯化直接用于下一步反应,收率95%。
(2) 化合物3的合成(以3a为例)
将4.75 mmol化合物2a加入反应瓶中,加入丙酮溶解(0.1 M),然后加入NaN3(5.70 mmol, 1.20 equiv),于室温下搅拌反应24 h。反应结束后,减压蒸馏除去溶剂,加入20 mL二氯甲烷(DCM)溶解有机物,再加入水(3×20 mL)洗涤。无水硫酸镁干燥有机相,过滤。减压蒸馏除去溶剂,得到的黄色固体为2-叠氮-1,4-萘醌(3a)不经进一步纯化直接用于下一步反应,收率92%。
(3) 化合物4的合成(以4a为例)
将4.37 mmol化合物3a加入反应瓶中,并用DCM(0.1 M)溶解,再称取PPh3(4.56 mmol, 1.05 equiv),并用DCM(1.0 M)溶解后,将该溶液滴加入反应瓶中,反应体系有大量气泡产生,约30 min后基本反应完全。反应结束后,通过二氯甲烷/石油醚重结晶以75%收率得到目标产物2-三苯基膦亚胺叶立德-1,4-萘醌(4a),洗涤固体的滤液再通过Al2O3柱层析得到产物4a,收率13%。
2-三苯基膦亚胺叶立德-1,4-萘醌[17](4a):红色固体,收率88%, m.p.173~174 ℃;1H NMR(400 MHz, Chloroform-d)δ: 8.01(d,J=7.7 Hz, 1H), 7.86(d,J=7.6 Hz, 1H), 7.81~7.75(m, 6H), 7.59(d,J=7.6 Hz, 1H), 7.54~7.45(m, 10H), 6.31(s, 1H);13C NMR(101 MHz, Chloroform-d)δ: 184.6(d,J=3.3 Hz), 183.6(d,J=14.6 Hz), 156.9(d,J=4.9 Hz), 133.9, 133.6, 132.5(d,J=9.8 Hz), 132.0(d,J=2.9 Hz), 131.6, 131.4, 129.7(d,J=102.9 Hz), 128.7(d,J=12.3 Hz), 126.4, 125.5, 116.5(d,J=19.4 Hz);31P NMR(162 MHz, Chloroform-d)δ: 13.2; HR-MS(ESI)m/z: calcd for C28H21NO2P+{[M+H]+}434.1310, found 434.1314。
2-三苯基膦亚胺叶立德-6,7-二甲基-1,4-萘醌(4b):红色固体,收率84%, m.p.177~178 ℃;1H NMR(400 MHz, Chloroform-d)δ: 7.81~7.75(m, 7H), 7.59(s, 1H), 7.54~7.49(m, 3H), 7.48~7.43(m, 6H), 6.29(s, 1H), 2.31(s, 3H), 2.26(s, 3H);13C NMR(101 MHz, Chloroform-d)δ: 185.1(d,J=3.2 Hz), 183.5(d,J=14.2 Hz), 143.3, 140.6, 132.4(d,J=9.9 Hz), 131.9(d,J=2.9 Hz), 130.5, 129.5, 129.4, 128.6(d,J=12.6 Hz), 127.6(d,J=91.3Hz), 116.4(d,J=20.0 Hz), 20.1, 19.7;31P NMR(162 MHz, Chloroform-d)δ: 12.9; HR-MS(ESI):m/zcalcd for C30H25NO2P+{[M+H]+}462.1623, found 462.1627。
2-三苯基膦亚胺叶立德-5,8-二甲基-1,4-萘醌(4c):红色固体,收率87%, m.p.172~174℃;1H NMR(400 MHz, Chloroform-d)δ: 7.78(dd,J=12.3 Hz, 7.6 Hz, 6H), 7.53~7.49(m, 3H), 7.47~7.43(m, 6H), 7.23(d,J=7.9 Hz, 1H), 7.11(d,J=7.9 Hz, 1H), 6.27(s, 1H), 2.69(s, 3H), 2.28(s, 3H);13C NMR(101 MHz, Chloroform-d)δ: 187.8(d,J=3.5 Hz), 186.0(d,J=13.5 Hz), 138.5(d,J=103.8 Hz), 137.1, 134.6, 132.5(d,J=9.8 Hz), 132.4, 131.7(d,J=2.9 Hz), 130.94, 130.9, 129.9, 128.5(d,J=12.3 Hz), 116.8(d,J=20.7 Hz), 23.1, 22.7;31P NMR(162 MHz, Chloroform-d)δ: 13.6; HR-MS(ESI):m/zcalcd for C30H25NO2P+{[M+H]+}462.1623, found 462.1617。
2-三苯基膦亚胺叶立德-5,6-二甲基-1,4-苯醌(4d):红色固体,收率72%, m.p.163~166 ℃;1H NMR(400 MHz, Chloroform-d)δ: 7.79~7.70(m, 6H), 7.57~7.5(m, 3H), 7.48~7.42(m, 6H), 5.94(s, 1H), 1.96(s, 3H), 1.85(s, 3H);13C NMR(101 MHz, Chloroform-d)δ: 187.4(d,J=2.7 Hz), 185.6(d,J=15.7 Hz), 154.7(d,J=4.7 Hz), 142.4, 137.0, 132.5(d,J=9.8 Hz), 131.9(d,J=2.9 Hz), 129.7(d,J=102.5 Hz), 128.7(d,J=12.3 Hz), 113.0(d,J=18.8 Hz), 77.4, 77.0, 76.7, 12.6, 12.3;31P NMR(162 MHz, Chloroform-d)δ: 12.5; HR-MS(ESI)m/z: calcd for C26H23NO2P+{[M+H]+}412.1467, found 412.1466。
2-三苯基膦亚胺叶立德-1,4-蒽醌(4e):红色固体,收率82%, m.p.212~215 ℃;1H NMR(400 MHz, Chloroform-d)δ: 8.50(s, 1H), 8.38(s, 1H), 7.96(d,J=8.0 Hz, 1H), 7.89(d,J=8.1 Hz, 1H), 7.84~7.78(m, 6H), 7.55~7.45(m, 11H), 6.50(s, 1H);13C NMR(101 MHz, Chloroform-d)δ: 184.2(d,J=3.2 Hz), 183.1, 157.9, 134.6(d,J=129.0 Hz), 133.5, 132.5(d,J=9.9 Hz), 132.0(d,J=2.9 Hz), 130.4, 129.8, 129.7, 128.9, 128.7(d,J=12.4 Hz), 128.688, 128.1, 126.8, 118.7(d,J=20.9 Hz);31P NMR(162 MHz, Chloroform-d)δ: 18.6; HR-MS(ESI)m/z: calcd for C32H23NO2P+{[M+H]+}484.1466, found 484.1457。
2 结果与讨论
2.1 反应条件优化
文献调研发现,当使用叠氮化钠作为叠氮化试剂时,通常使用N,N-二甲基甲酰胺和二甲基亚砜作为反应溶剂。但由于N,N-二甲基甲酰胺和二甲基亚砜均为高沸点溶剂,在后处理过程中难以完全除去,不利于反应放大,因此在合成化合物3a的过程中,一系列低沸点的反应溶剂被筛选(表1),结果发现,当使用丙酮作溶剂时,能以92%收率得到叠氮化产物3a。此外,产物4a的稳定性较差,易水解生成2-氨基-1,4-萘醌和三苯基氧膦,且随着反应时间的延长,收率下降明显,当在反应体系中加入2.00 equiv无水硫酸镁后,反应收率得到了明显提高(表2)。

表1 第2步反应条件优化aTable 1 Condition optimization of the second step reaction

表2 第3步反应条件优化aTable 2 Condition optimization of the third step reaction
2.2 底物扩展和反应放大
在最佳反应条件下,6,7-二甲基-1,4萘醌(1b)和5,8-二甲基-1,4-萘醌(1c)均能以良好收率得到最终的目标化合物4b和4c(图3)。1c可能由于目标产物4d的共轭性减弱,使4d稳定性减弱,反应副反应增加,收率降低明显。此外,1,4-蒽醌(1e)也能适用于该反应,能以69%总收率得到目标产物4e。为了考察该合成路线的放大效果,初始原料1a的用量被增加至30 mmol时,仍然能以74%总收率得到目标产物4a,说明该合成路线具有实际应用价值。
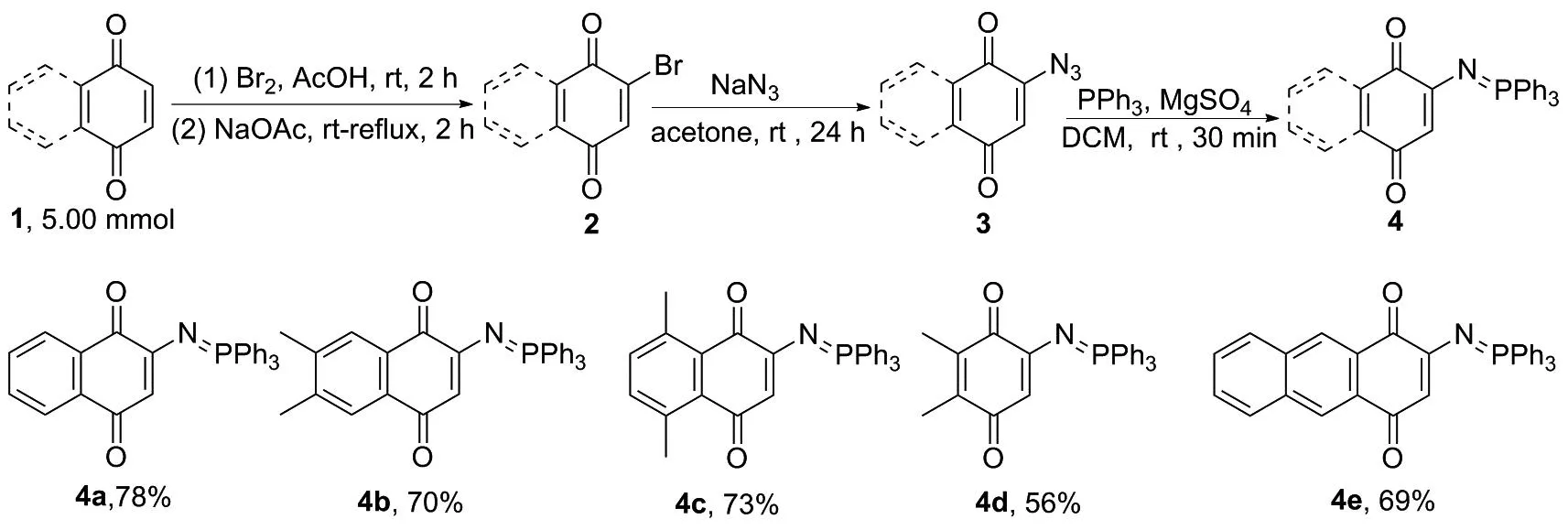
图3 底物扩展Figure 3 Scope of substrates
本文利用1,4-醌类化合物为原料,通过溴代反应、叠氮化反应和Staudinger反应,以中等到良好的收率合成了5种2-膦亚胺-1,4-醌类化合物。其中化合物4b~4e为新型化合物。该合成路线操作简单,副反应少,适合放大,具有实际应用的前景。
