3 种常用碳青霉烯类抗生素血药浓度UPLC-MS/MS 检测方法的建立 Δ
2024-02-28张瑞霞吕雅瑶翁莉莉天津医科大学一中心临床学院天津3009天津市第一中心医院药学部天津3009
秦 怡 ,张瑞霞 ,吕雅瑶 ,翁莉莉 ,张 弋 (.天津医科大学一中心临床学院,天津 3009;.天津市第一中心医院药学部,天津 3009)
碳青霉烯类抗生素具有抗菌谱广、抗菌活性强、耐药率低的特点,已成为治疗重症感染的主要选择。最佳的抗生素剂量、适当的抗生素浓度和足够的给药疗程是重症感染治疗成功的关键[1]。碳青霉烯类抗生素多为亲水性药物,主要以原型形式通过肾脏排泄,故其易受肾脏清除率和分布容积的影响[2]。半数以上的重症患者在使用与非重症患者剂量相同的抗生素给药时,会出现剂量不足的现象,造成抗生素耐药和感染控制无效,从而增加患者的住院时间,甚至降低患者的生存质量[3]。此外,碳青霉烯类抗生素的蓄积还会导致神经毒性[4]。目前,治疗药物监测的目的已逐步从避免药物毒性反应转移为确保疗效和减少治疗指数较宽药物的不良反应[5—6]。《重症患者β-内酰胺类抗生素治疗的优化指南(2019 版)》明确指出,使用治疗药物监测可提高β-内酰胺类抗生素药动学/药效学目标实现的达标率,降低药物处于次优浓度和发生剂量相关不良反应的风险[7]。
目前,我国临床用碳青霉烯类抗生素主要为亚胺培南(imipenem,IPM)、美罗培南(meropenem,MEM)、厄他培南(ertapenem,ETP),通常为单一用药。然而有研究表明,双碳青霉烯联合疗法(ETP 联合MEM)可以增强碳青霉烯类药物对耐药肠杆菌/肺炎克雷伯菌的抗菌作用,用于多黏菌素类抗生素导致肾功能受损的重症感染患者[8—9]。由于临床用药方案的改进及血浆中碳青霉烯类抗生素的不稳定性[10],故需满足临床上重症患者不同碳青霉烯类抗生素血药浓度定量需求,即定量方法应达到同时快速定量多种药物血药浓度的要求。超高效液相色谱-质谱联用(ultra-high performance liquid chromatography-mass spectrometry,UPLC-MS/MS)法灵敏度和特异度高,操作简单快速,适用于多种药物监测。本研究建立了一种简单快速测定3种常用碳青霉烯类抗生素(ETP、IPM、MEM)血药浓度的UPLC-MS/MS法,用于监测重症患者的血药浓度,以期为患者个体化给药提供参考。
1 材料
1.1 主要仪器
Waters ACQUITY UPLC Ⅰ-Class 系统、Xevo TQD系统、Masslynx V4.2 工作站均购自美国Waters 公司;MX-F 型固定式涡旋混合仪购自美国Scilogex 公司;BY-G20 型高速离心机购自北京白洋医疗器械有限公司;SB-5200DT型超声波清洗机购自宁波新芝生物科技有限公司。
1.2 主要药品与试剂
ETP(纯度98.16%,批号V-806800-NU1)、IPM(纯度98.14%,批号V-804800-NU1)、MEM(纯度86.80%,批号130506-202004)均购自中国食品药品检定研究院;ETPD4(内标,同位素丰度99.00%,批号12-MRS-161-1)购自上海甄准生物科技有限公司;IPM-D4(内标,同位素丰度99.50%,批号7-MMH-92-2)、3-吗啉丙磺酸(3-morpholinopropanesulfonic acid,MOPS)缓冲剂购自美国CFW公司;MEM-D6(内标,同位素丰度99.70%,批号5-JUZ-42-1)购自谱析(上海)生物科技有限公司。甲醇、乙腈均购自美国Thermo Fisher Scientific公司;甲酸购自上海安谱科学仪器有限公司;水为屈臣氏蒸馏水。
1.3 人血浆
血浆为健康者未用药的空白血浆。本研究获天津医科大学一中心临床学院伦理委员会审批,伦理号为科研20240124-1。
2 方法与结果
2.1 色谱与质谱条件
色谱柱为ACQUITY UPLC BEH C18(2.1 mm×50 mm,1.7 μm);流动相A 为98%水+2%乙腈+0.1%甲酸,流动相B 为98%乙腈+2%水+0.1%甲酸,梯度洗脱(0~0.50 min,95%A;0.50~0.51 min,95%A→85%A;0.51~1.60 min,85%A→65%A;1.60~1.70 min,65%A→95%A;1.70~3.50 min,95%A);流速为0.3 mL/min;进样量为8 μL;柱温为40 ℃;进样器为8 ℃。离子源为电喷雾离子源,采用多反应监测(multiple reaction monitoring,MRM)方式,检测模式为正离子模式。ETP、IPM、MEM及其内标的质谱检测参数见表1。
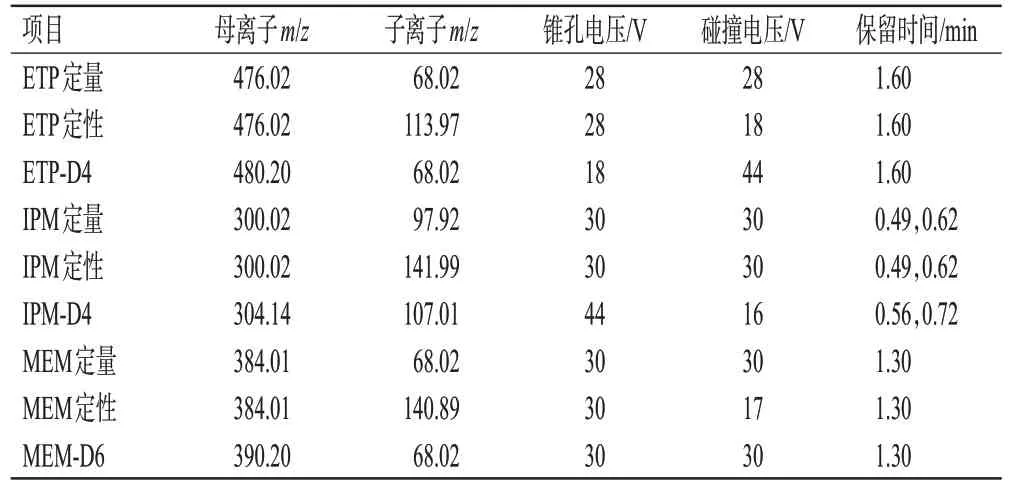
表1 ETP、IPM、MEM及其内标的质谱检测参数
2.2 相关溶液与血浆样品的配制
2.2.1 稳定剂配制
精密称取MOPS 缓冲剂10.46 g,置于250 mL 容量瓶中,加纯水溶解并定容,得0.2 mol/L MOPS缓冲液,用1 mol/L NaOH 调节pH 至6.8,即得稳定剂,置于4 ℃冰箱冷藏备用。
2.2.2 ETP、IPM、MEM标准品溶液配制
精密称取ETP、IPM、MEM 各1.00 mg,分别置于EP管中,加“2.2.1”项下稳定剂250 μL 溶解,配制成质量浓度均为4 000 μg/mL 的标准品储备液。将上述适量ETP、IPM、MEM 储备液以2∶1∶1 比例混匀,得质量浓度分别为2 000、1 000、1 000 μg/mL 的混合标准品溶液。取混合标准品溶液,用稳定剂倍比稀释,得ETP 质量浓度分别为2 000、1 000、200、100、20、10、5、2 μg/mL,IPM、MEM质量浓度分别均为1 000、500、100、50、10、5、2.5、1 μg/mL 的混合系列标准工作液。同时,用稳定剂配制ETP质量浓度为1 600、100、5 μg/mL,IPM、MEM质量浓度分别均为800、50、2.5 μg/mL 的混合质控样品溶液。各标准品储备液于-80 ℃避光保存,系列标准工作液和混合质控样品溶液现配现用。
2.2.3 内标溶液配制
精密称取ETP-D4、IPM-D4、MEM-D6各1.00 mg,分别置于5 mL 棕色容器中,加稳定剂溶解并定容,摇匀,配制成ETP-D4、IPM-D4、MEM-D6 质量浓度均为200 μg/mL的内标储备液。取适量储备液,加稳定剂稀释得ETP-D4、IPM-D4、MEM-D6 质量浓度分别为25、100、25 μg/mL的混合内标溶液,分装后于-80 ℃避光保存备用。
2.2.4 混合标准曲线样品和质控血浆样品的配制
精密吸取180 μL空白血浆,置于1.5 mL离心管中,加入ETP、IPM、MEM 混合系列标准工作液20 μL,涡旋15 s 混匀,得ETP 质量浓度为200、100、20、10、2、1、0.5、0.2 μg/mL,IPM、MEM质量浓度分别均为100、50、10、5、1、0.5、0.25、0.1 μg/mL的混合标准曲线样品。混合步骤同标准曲线样品,配制成ETP 质量浓度为160、10、0.5 μg/mL,IPM、MEM质量浓度分别均为80、5、0.25 μg/mL的高、中、低浓度的混合质控血浆样品溶液。
2.3 血浆样品预处理
精密吸取100 μL 血浆样品,与稳定剂1∶1 混合,加入10 μL 混合内标溶液(ETP-D4 为25 μg/mL,IPM-D4为100 μg/mL,MEM-D6 为25 μg/mL),涡旋15 s;加入400 μL甲醇,涡旋30 s,以13 000 r/min离心10 min;取上清液,用0.22 μm微孔滤膜过滤,取滤液100 μL,加超纯水300 μL,涡旋10 s,以13 000 r/min离心10 min后取上清液,进样。
2.4 方法学验证
2.4.1 专属性
分别取健康者的空白血浆加入中浓度的混合质控样品溶液和混合内标溶液,均按“2.3”项下方法处理,记录色谱图。结果显示,ETP、IPM、MEM与对应内标色谱峰峰形良好,完全分离。ETP 和ETP-D4 保留时间均为1.60 min;MEM和MEM-D6保留时间均为1.30 min;IPM和IPM-D4以双峰存在,其中IPM双峰保留时间为0.49、0.62 min,IPM-D4 双峰保留时间为0.56、0.72 min。人血浆中内源性物质不干扰药物和对应内标的测定,该方法专属性良好。结果见图1(空白血浆图略)。
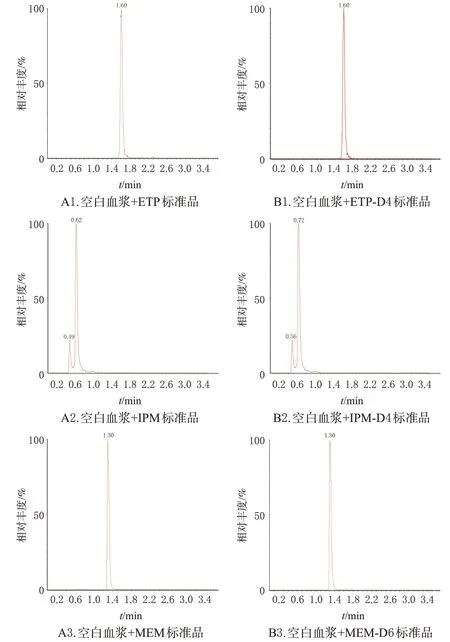
图1 ETP、IPM、MEM及其内标的MRM图谱
2.4.2 标准曲线与定量下限
取ETP、IPM、MEM混合标准曲线样品,每一质量浓度各3 份,按“2.3”项下方法处理,记录峰面积。分别以对照品质量浓度为横坐标(X),3种药物与其内标的峰面积比值为纵坐标(Y),采用W=1/X2进行线性回归运算,得到ETP、IPM、MEM 在0.2~200、0.1~100、0.1~100 μg/mL范围内线性良好,线性方程分别为Y=2.877 67X-0.082 915(r2=0.997)、Y=4.195 77X-0.167 327(r2=0.993)、Y=6.256 62X-0.074 893(r2=0.994)。以最低浓度为定量下限(lower limit of quantification,LLOQ)。取6 份ETP、IPM、MEM 质量浓度为0.2、0.1、0.1 μg/mL的混合标准曲线样品,按照“2.3”项下方法处理后记录实测浓度。实测浓度的相对误差(relative error,RE)<3.60%,相对标准偏差(relative standard deviation,RSD)<9.41%。
2.4.3 提取回收率与基质效应
取不同来源空白血浆配制低(ETP 为0.5 μg/mL,IPM、MEM分别均为0.25 μg/mL)、中(ETP为10 μg/mL,IPM、MEM 分别均为5 μg/mL)、高(ETP 为160 μg/mL,IPM、MEM 分别均为80 μg/mL)3 种质量浓度的质控样品,按“2.3”项下方法处理,每个质量浓度平行制备6份,将对照品与内标峰面积的比值记为A。取6份不同来源的空白血浆按“2.3”项下方法处理后,加入高、中、低3种质量浓度混合质控样品溶液和混合内标溶液,对照品和内标峰面积比值记为B。取6 份低(ETP 为5 μg/mL,IPM、MEM分别均为2.5 μg/mL)、中(ETP为100 μg/mL,IPM、MEM分别均为50 μg/mL)、高(ETP为1 600 μg/mL,IPM、MEM分别均为800 μg/mL)3种质量浓度的混合质控样品溶液10 μL,与190 μL稳定剂、400 μL甲醇混匀,以超纯水适当稀释后上样检测,对照品和内标峰面积比值记为C。提取回收率(%)=A/B×100%,基质效应(%)=B/C×100%。结果显示,ETP、IPM、MEM 低、中、高3种质量浓度混合质控样品内标归一化的提取回收率及基质效应较一致,全部RSD≤12.99%,详见表2。
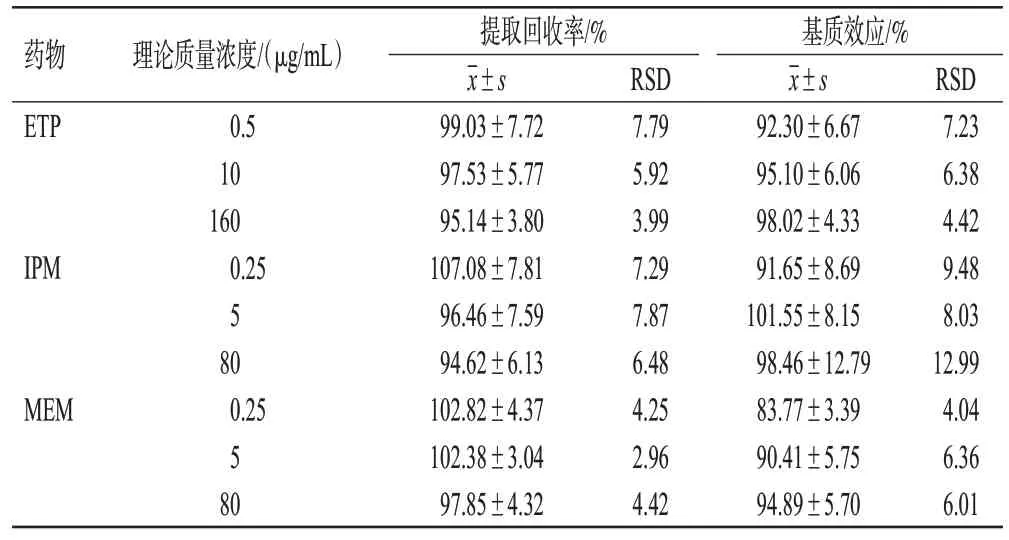
表2 ETP、IPM、MEM 混合质控样品内标归一化的提取回收率及基质效应(n=6)
2.4.4 精密度和准确度
配制ETP 质量浓度分别为0.2、0.5、10、160 μg/mL,IPM、MEM 质量浓度分别均为0.1、0.25、5、80 μg/mL 的LLOQ 和低、中、高质量浓度的混合血浆标准样品,按“2.3”项下方法处理,每个质量浓度平行制备5 份,连续3 d重复测定,根据随行标准曲线计算样品的实测浓度。ETP、IPM、MEM的LLOQ和低、中、高质量浓度的批内、批间RE均≤5.14%、RSD均≤11.15%,详见表3。

表3 ETP、IPM、MEM 在血浆中的批内及批间精密度和准确度(n=5)
2.4.5 稳定性
待测物低、中、高3个质量浓度的混合质控样品溶液与血浆混合后,分别在室温放置4、8、12、24 h 和4 ℃放置4、8、12、24 h 考察血浆样品短期稳定性,在-80 ℃冰箱冻存60 d考察血浆样品长期稳定性;冻融循环3次考察血浆样品冻融稳定性。血浆样品按“2.3”项下方法处理后置于处理进样器(8 ℃)和室温4、8、12、24 h,室温放置4、8、12、24 h,考察处理后血浆样品稳定性。4 000 μg/mL的ETP、IPM、MEM 标准品储备液分装,置于-80 ℃冻存60 d,考察储备液稳定性。RE 和RSD 均<15%视为稳定性良好。结果显示,ETP、IPM、MEM血浆样品在室温条件下分别可以稳定8、4、8 h,在4 ℃下分别可以稳定24、8、24 h,在-80 ℃冰箱冻存60 d、冻融循环3 次稳定性良好。ETP、IPM、MEM 血浆样品处理后在进样器(8 ℃)中分别可以稳定24、4、24 h,室温条件下分别可以稳定8、4、8 h。ETP、IPM、MEM标准品储备液在-80 ℃冰箱冻存60 d可以保持稳定。
3 讨论
3.1 内标物质的选择
稳定性同位素内标是UPLC-MS/MS 定量分析方法的首选,本实验选择ETP-D4、IPM-D4、MEM-D6 这3 种同位素内标,分别与3种待测物ETP、IPM、MEM一一对应。同位素内标与待测物的理化性质完全一致,不仅可以消除仪器进样的误差,还在最大程度上消除了基质效应。本实验根据调谐结果分别比较了4 对IPM-D4 离子对(304.14→107.01、304.14→141.98、304.07→97.91、304.07→106.96)的质谱监测参数和响应强度。结果显示,304.14→141.98、304.07→97.91 这2 组保留时间与IPM 一致,但其信噪比和响应强度均不佳;304.14→107.01 虽与IPM 保留时间不一致,但其峰面积位于IPM线性范围中段且线性范围良好。
3.2 血浆样品与前处理方法的选择
研究显示,与血清、柠檬酸盐乙二胺四乙酸血浆样品相比,肝素化血浆样品中IPM 和MEM 质量浓度超过标称值的115%,低浓度尤其明显,故本实验血样采集选择柠檬酸盐乙二胺四乙酸抗凝管[4]。生物样品前处理方法常用蛋白沉淀法、液-液萃取法、固相萃取法等,碳青霉烯类抗生素较不稳定,简单、高效的前处理方法可最大限度地缩短样品周转时间,因此本实验选择了操作简单的蛋白沉淀法。本实验前处理中比较了以甲醇(分别为3∶1、4∶1、5∶1)和乙腈(分别为2∶1、3∶1、4∶1)沉淀血浆样品蛋白的6种方式。结果发现,乙腈沉淀血浆样品蛋白时,无论其比例如何,ETP 均出现裂峰,IPM、MEM 出现不同程度的分峰或拖尾。以甲醇3∶1 沉淀血浆样品蛋白时,IPM分峰;以甲醇5∶1沉淀血浆样品蛋白时,IPM 峰展宽,ETP 出现前沿峰;以甲醇4∶1 沉淀血浆样品蛋白时,ETP、IPM、MEM 峰形和响应强度均较好,同时蛋白沉淀效果良好,故本研究最终选择甲醇4∶1沉淀血浆样品蛋白。沉淀蛋白后上清液与流动相初始梯度极性相差较大,直接上样后ETP、IPM、MEM不同程度地出现拖尾峰。为确保分析物峰形良好并降低基质效应,上样前应加水稀释上清液,且当上清液与水的比例为1∶3时,化合物的峰形、线性范围、基质效应、提取回收率均良好。
3.3 碳青霉烯类抗生素稳定性分析
碳青霉烯类抗生素,尤其是IPM 在血浆中极不稳定,并且会受到浓度与温度的影响[10]。兼性离子缓冲液可延缓碳青霉烯类抗生素的降解[11—12],故本实验选择0.2 mol/L MOPS 作为稳定剂。但是MOPS 可能会损伤色谱柱,干扰方法稳定性,并且会增加检测成本[13]。所以本实验只在保存样品与制备样品时加入稳定剂,不在流动相中加入稳定剂。此外,β-内酰胺类抗生素环状结构对pH 敏感,当pH>6 时,会产生降解,且碱性越强降解越快。因此,本实验在流动相的水相和有机相中加入0.1%甲酸调节pH,使流动相pH 为酸性,同时避免碱性物质加入,减少药物降解[14]。
临床上若对碳青霉烯类抗生素进行治疗药物监测,应重视取血到上机分析时的保存条件和温度。此外,本实验发现,将IPM置于4 ℃保存12 h或冻融循环3次后,仅高浓度组不满足稳定性要求(RE>15%)。以往研究表明,更高的温度与药物浓度会导致IPM注射液稳定性降低,故临床上检测IPM 血药浓度应倾向选择稳态谷浓度[10]。
3.4 IPM双峰现象
IPM 已被证明在水溶液中存在顺式和反式两种形式的构象异构体[4]。IPM的两种构象异构体处于平衡状态,可以通过UPLC 分离[15—16]。本实验中IPM 以稳定的双峰形式存在,未发生展宽或者合并现象,且基线未完全分离,因此根据双峰的总面积进行定量。此外,本实验用稳定同位素内标IPM-D4矫正了在测量浓度范围内IPM 的峰形变化及与IPM 共洗脱极性内源性化合物的基质效应。
综上所述,本实验建立了一种可以同时定量临床3种常用碳青霉烯类抗生素(ETP、IPM、MEM)血药浓度的UPLC-MS/MS 法,该方法样品前处理简单,检测时间短,所需样品量少,可以满足临床上(尤其重症患者)对碳青霉烯类抗生素血药浓度的监测需求。
