氮掺杂二硫化钼纳米催化剂的电催化析氢性能
2024-02-23杨成功黄蓉王冬娥田志坚
杨成功,黄蓉,王冬娥,田志坚
(1 中国科学院大连化学物理研究所,辽宁 大连 116023;2 中国科学院大学,北京 100049)
随着世界各国对碳排放提出更严格的要求,特别是我国提出“碳达峰”和“碳中和”的发展目标以应对日益严重的气候变化问题,低碳清洁能源将逐渐成为未来世界能源需求的主要组成部分[1]。氢能具有高的能量密度,燃烧的产物是水,这使其成为未来清洁能源的最佳选择[2]。从太阳能、风能等其他可再生能源中获得电能,然后电解水产氢被认为是一种清洁、可持续的产氢途径[3-6]。电解水产氢的基本反应为阴极上水的还原[析氢反应(HER):2H++2e-→H2][7]。在此过程中需要催化剂以降低HER 的能垒,降低反应过电位。因具有最适宜的氢原子吸附吉布斯自由能(ΔGH*=0),Pt 族贵金属被认为是最高效的HER催化剂[8]。但高昂的成本限制了Pt催化剂在实际中的广泛应用。因此,开发廉价高效的HER 催化剂是电催化制氢产业化的当务之急。
近年来的研究发现,储量丰富、成本低廉的二硫化钼(MoS2)具有出色的电催化性能,成为新型的电催化产氢材料[9-11]。MoS2作为一种典型的二维层状材料,其片层边缘位点具有HER 催化活性,而基面是惰性的[12]。然而,在MoS2材料中,惰性的基面占比较大,活性边缘位点暴露量有限,限制了其潜在的HER催化性能[10,13]。近年来,研究人员发现,化学掺杂可以将原子掺杂进MoS2晶格中,使得掺杂原子附近Mo 和S 原子的电子密度受到掺杂原子的显著影响,产生新的不饱和配位点,从而提高MoS2催化剂的HER活性[14-17]。
化学掺杂包括金属掺杂和非金属掺杂。金属掺杂通常是将Co、Ni、Fe、Cu 和V 等金属原子掺杂进MoS2的晶格[8]。例如,Co、Ni 掺杂MoS2形成的“Co(Ni)MoS”相被认为是催化活性位[18]。Co掺杂使得MoS2表面生成大量不饱和配位点,从而提高MoS2的HER 催化活性[19]。此外,还可以通过非金属掺杂来提高MoS2的催化活性。非金属掺杂通常是将O、N、P 和B 等非金属原子掺杂进MoS2晶格[8]。Xie 等[16]报道O 掺杂可以降低MoS2的结晶度,增加不饱和配位点的数量,从而提高MoS2的HER催化活性。Liu 等[20]发现P 掺杂MoS2可以在惰性的基面上生成新的不饱和配位点,从而表现出优异的HER 催化活性。N 掺杂剂也已经被应用于MoS2电子结构的调节,并将N 掺杂改性的MoS2催化剂应用于电催化还原CO2[21]和电催化还原N2制备NH3[22]。N是一种非常有应用前景的掺杂剂,可以稳定掺杂进MoS2中,并改善其电化学性能。
在本研究中,以钼酸钠为钼源,L-半胱氨酸为硫源和还原剂,以双氰胺为氮源,采用水热法合成了氮掺杂二硫化钼(N-MoS2)催化剂。通过SEM 和XPS 等手段分别表征了其形貌和电子性质。采用电化学工作站在酸性介质中测试了催化剂的线性扫描伏安曲线和塔菲尔斜率,评价了N-MoS2纳米催化剂的电催化析氢性能。
1 实验材料和方法
1.1 材料
钼酸钠(Na2MoO4,分析纯),光华化学试剂厂;L-半胱氨酸(分析纯),上海阿拉丁试剂公司;双氰胺(C2H4N4,分析纯),上海阿拉丁试剂公司。
1.2 N-MoS2催化剂的制备
将10mmol (2.42g) Na2MoO4∙2H2O、40mmol(4.84g)L-半胱氨酸和一定量的双氰胺(C2H4N4)溶于60mL 去离子水中得到均匀溶液。然后,将溶液转移到带有聚四氟乙烯内衬的不锈钢反应釜中,在180℃的烘箱中水热24h,冷却到室温。最后,将固体产物过滤,用去离子水和乙醇洗涤3 次,60℃真空烘箱中干燥12h,得到氮掺杂的MoS2纳米催化剂(N-MoS2)粉末。通过调变双氰胺的添加量,使双氰胺中N 与钼酸钠中Mo 的原子比分别为0.05、0.1 和0.2,得到的样品分别命名为N-MoS2-0.05、N-MoS2-0.1和N-MoS2-0.2。作为对照,MoS2催化剂的合成过程与N-MoS2相同,但不添加双氰胺。
1.3 N-MoS2催化剂的表征
采用配备X 射线能谱仪的场发射扫描电镜(FESEM,型号JSM-7800F)观察材料的形貌和高分辨结构。并用与扫描电镜配套的X-max 80 硅漂移探测器进行元素分布分析(mapping)。采用红外光谱仪(型号Thermo Fisher Nicolet iS50)采集了样品的红外光谱(FTIR spectra)。采用X’Pert PRO(PANalytical)衍射仪记录样品的X射线衍射(XRD)图谱,Cu Kα作为射线发射源(λ=0.15418nm),管电压为40kV,管电流为40mA,扫描范围为5°~90°。采用共聚焦拉曼显微镜(型号Renishaw inVia)采集了样品的拉曼光谱,激发波长532nm。使用X 射线光电子能谱(XPS,型号Thermo Fisher ESCALAB 250xi)分析材料的组成和化学结构,其中X 射线发射源为Al Kα,用C 1s 的电子结合能(284.6eV)为标准进行结合能校正。采用有机元素分析仪(型号Flash 2000,Thermo Fisher Scientific)进行了有机元素分析测试。采用电感耦合等离子体发射光谱仪(ICP-OES,型号7300DV,PerkinElmer)测定了样品中Mo元素的含量。
1.4 N-MoS2催化剂的HER性能评价
HER 性能评价是在典型的三电极系统中用CHI660E电化学工作站完成的。对电极为铂丝,参比电极为饱和Hg/HgCl电极。所有测量均在N2饱和的0.5mol/L H2SO4水溶液中进行。工作电极的制备:将4mg 催化剂、35.8μL Nafion 与2mL 乙醇,超声分散均匀后,取7.2μL 滴涂在直径3mm 的玻璃碳电极表面,晾干成膜,以保证催化剂的负载量为0.2mg/cm2。采用电化学工作站测量和研究工作电极的电催化析氢性能,包括线性扫描伏安曲线(LSVs)和塔菲尔(Tafel)曲线。
2 结果与讨论
图1 为合成的MoS2和N-MoS2催化剂的XRD 图谱。图中14.2°、32.2°、35.6°和57.2°出现的衍射峰分别对应2H-MoS2的(002)、(100)、(102)和(110)的特征峰,且所有样品的XRD 图谱均符合2H-MoS2的标准粉末衍射图谱(JCPDS No.75-1539)。水热过程中,Na2MoO4中的Mo6+被L-半胱氨酸还原成Mo4+,并被L-半胱氨酸上的巯基硫化生成S-Mo-S原子层,结晶长大生成MoS2。与此同时,Mo4+还可以与双氰胺分解生成的氨反应生成Mo-N,使得N掺杂进MoS2晶体。相较于MoS2催化剂,N-MoS2催化剂并没有产生新的衍射峰,表明N掺杂并没有改变MoS2的晶相结构。局部放大的XRD图谱显示NMoS2的(002)主峰相对于MoS2催化剂发生了低角度位移。根据布拉格方程可知,对应的N-MoS2样品的层间距也大于MoS2样品。XRD 图谱显示NMoS2催化剂的(002)衍射峰较MoS2样品变宽,衍射峰强度降低,表明N 掺杂使得MoS2催化剂的结晶度降低,具有更多的无序结构[23-25]。

图1 MoS2和N-MoS2催化剂的X射线衍射图
图2 为MoS2和N-MoS2催化剂的SEM 图,以及N-MoS2-0.1的元素mapping图。SEM结果显示,所有样品均为纳米片组成的花球。N 元素的掺杂对MoS2的形貌没有明显影响。扫描电镜下进行的元素mapping 显示N-MoS2-0.1 样品中除了Mo 和S 元素之外,还含有N元素,且所有元素分布均具有良好的空间对应关系,N 元素在N-MoS2-0.1 样品中均匀分布。

图2 MoS2和N-MoS2催化剂的SEM图和N-MoS2-0.1的元素mapping图
图3 为MoS2和N-MoS2催化剂的XPS 图谱,给出了Mo 3d、S 2s和N 1s的结合能。位于228.8eV和231.9eV 的特征峰分别对应于Mo4+的Mo 3d 5/2和Mo 3d 3/2[26],位于225.9eV 的特征峰为S 2s 的峰[27]。位于161.3eV 和162.6eV 的特征峰分别为S2-的S 2p3/2和S 2p1/2[28]。位于349.5eV的特征峰为Mo 3p 3/2的峰[29]。相较于N-H 键中位于401~402eV 的N 1s 的特征峰[30],图3 中位于397.5eV 左右的特征峰出现负移,可归属为Mo-N 键中N 1s 的峰[31],表明NMoS2样品中的N以Mo-N键的形式存在。
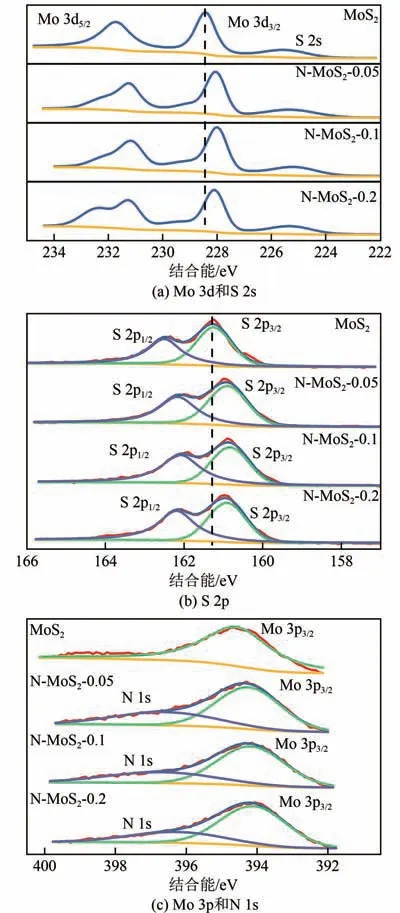
图3 MoS2和N-MoS2催化剂的XPS图谱
从图3可以看出,相较于MoS2催化剂,N-MoS2催化剂中Mo元素和S元素的结合能均出现了负移,N-MoS2-0.1催化剂中Mo和S元素结合能负移最大。表明Mo 原子和S 原子周围的电子结合能降低,可归结为Mo 原子和S 原子周围电子密度的增加[32-33]。电子密度的增加可以弱化Mo—S键,促进S空位的生成[34]。表1给出了由XPS得到的不同催化剂中S/Mo的原子比。结果显示,N-MoS2催化剂中S/Mo 原子比均小于MoS2的化学计量比2,表明N的掺杂使得MoS2生成了大量的S 空位,且N-MoS2-0.01 样品中的S空位最多。N是以取代S形成N-Mo的形式掺杂进MoS2晶格的,N-MoS2催化剂中(N+S)/Mo 原子比同样小于2,表明N的掺杂使得MoS2中存在不饱和配位Mo 原子,且N-MoS2-0.01 样品中的不饱和配位Mo 原子最多。此外,采用有机元素分析仪和ICP-OES 进行MoS2和N-MoS2催化剂中的N、S 和Mo 元素含量测定,并根据得到的各元素质量分数计算得出了N/Mo和S/Mo原子比。结果显示,在所有N-MoS2样品中均存在N、S、Mo元素。表2给出了元素分析和ICP-OES 得到的不同N-MoS2催化剂中N、S、Mo元素的质量分数以及N/Mo和S/Mo的原子比。表1中由XPS结果得到的N/Mo原子比表明,N-MoS2-0.05中N/Mo原子比约为0.05,N-MoS2-0.1中N/Mo 原子比约为0.10,而N-MoS2-0.2 中N/Mo原子比约为0.05。表2 中元素分析和ICP-OES 得到的N/Mo 原子比表明,N-MoS2-0.05 中N/Mo 原子比为0.06,N-MoS2-0.1 中N/Mo 原子比为0.10,而N-MoS2-0.2 中N/Mo 原子比为0.06。结果表明,由有机元素分析仪和ICP-OES 测试得到的N/Mo 和S/Mo 原子比结果与XPS 得到的结果基本一致,且S/Mo和(N+S)/Mo 原子比均小于2。元素分析结果进一步表明,N 的掺杂使得MoS2生成了大量的S 空位,且N-MoS2-0.01样品中的S空位最多。
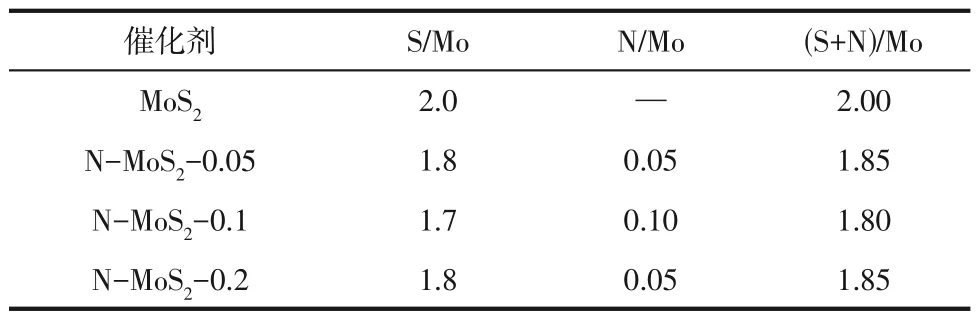
表1 XPS得到的MoS2和N-MoS2催化剂中的S/Mo和N/Mo原子比
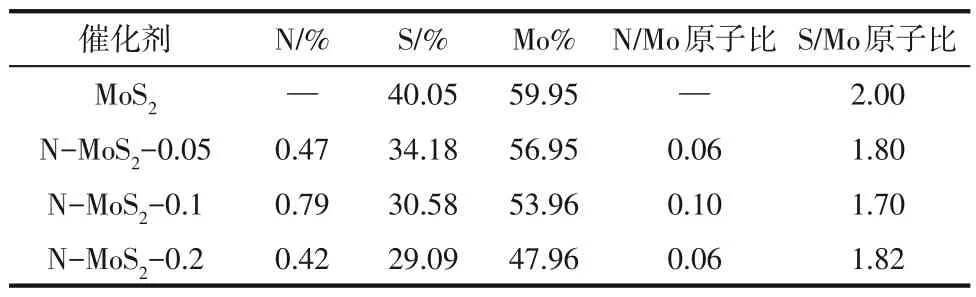
表2 元素分析和ICP-OES得到的催化剂中的N、S、Mo元素质量分数和原子比
其中N-MoS2-0.2 催化剂中测得的N/Mo 原子远小于投料比。这可能是由于N投料量较多时,N原子没有全部掺杂进MoS2晶格,多余的含N物质在样品制备过程中被洗去。根据文献报道,在纳米晶体中存在自净化效应,较大掺杂量时掺杂前后体系能量变化较大,会由于晶体的自净化效应使得杂原子难以掺杂进晶格或被排出晶格之外[35]。当含N原料投料量适中时,掺杂前后体系能量变化较小,N原子易于掺杂进MoS2晶格。含N原料投入较多时,大量的含N原料不仅会在局部聚集,且由于晶体的自净化效应而难以掺杂进MoS2晶格,甚至部分已经掺入MoS2晶格的N原子在MoS2晶化的过程中被排出,导致N-MoS2-0.2样品的实际掺杂量降低,甚至低于N-MoS2-0.1中的掺杂量。在金属掺杂MoS2的研究中也观察到过类似现象:过量的金属掺杂(Cu、Zn等)时只有少量可以进入到MoS2晶格中[14]。
图4为MoS2和N-MoS2催化剂的拉曼光谱。MoS2和N-MoS2催化剂在377.1和405.4cm-1处观察到对应于2H-MoS2的E1
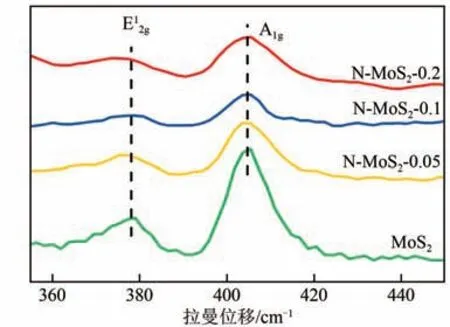
图4 MoS2和N-MoS2催化剂的拉曼光谱
2g和A1g拉曼峰。其中E12g为S-Mo-S原子层的面内剪切振动模式,A1g是S原子的面外振动模式[36]。N 的掺杂并没有产生新的拉曼特征峰,表明N 掺杂没有改变MoS2的层状结构。N-MoS2催化剂拉曼峰强度的弱化表明其结晶度降低,具有较多的无序结构。拉曼表征结果与XRD的结果一致。
图5 为MoS2和N-MoS2催化剂的线性扫描伏安曲线(LSVs)。通过线性扫描伏安测试得到的极化曲线给出了不同催化剂的电催化析氢过电位,可以比较同一电流密度时不同催化剂的过电位大小。从图中可以得出,在0.5mol/L H2SO4酸性介质中,当电流密度为10mA/cm2时,MoS2催化剂的过电位为512mV,N-MoS2-0.05 催化剂的过电位为363mV,N-MoS2-0.1催化剂的过电位为305mV,N-MoS2-0.2催化剂的过电位为365mV。在相同电流密度下,N-MoS2催化剂的过电位小于MoS2催化剂上的过电位,且N-MoS2-0.1 催化剂的过电位最小,更接近Pt催化剂。

图5 MoS2和N-MoS2催化剂的线性扫描伏安特性曲线
图6 给出了MoS2和N-MoS2催化剂的塔菲尔斜率。催化剂的塔菲尔斜率越小,催化活性越好[8]。MoS2催化剂的塔菲尔斜率为150mV/dec,N-MoS2催化剂的塔菲尔斜率在60~90mV/dec,均小于MoS2催化剂的塔菲尔斜率。N-MoS2-0.1 催化剂的塔菲尔斜率最小,为60mV/dec。以上电化学性能的结果表明,N 掺杂得到的N-MoS2催化剂的电催化析氢性能优于MoS2催化剂,且N-MoS2-0.1 催化剂的电催化析氢性能最优,更接近于Pt 催化剂的电催化析氢性能。
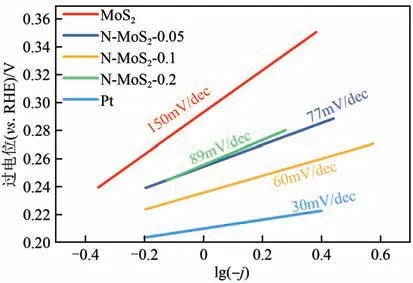
图6 MoS2和N-MoS2催化剂的塔菲尔曲线
在酸性介质中,析氢反应历程中电极表面的反应主要包括以下3种[37],如式(1)~式(3)。
电化学步骤(Volmer反应)

析氢反应主要通过以下两种路径进行[38]:①Volmer-Heyrovsky 路 径[式(1) 和 式(2)];②Volmer-Tafel 路径[式(1)和式(3)]。其中式(1)Volmer 反应是两种路径的共同步骤。通过极化曲线测量得到的塔菲尔曲线的斜率可以判断反应路径和速控步骤[39]:①塔菲尔斜率大于120mV/dec 时,析氢反应通过Volmer-Heyrovsky路径进行,速控步骤为式(1)Volmer反应;②塔菲尔斜率为40~120mV/dec时,析氢反应通过Volmer-Heyrovsky路径进行,速控步骤为式(2)Heyrovsky 反应;③塔菲尔斜率在30mV/dec 左右时,析氢通过效率最高的Volmer-Tafel路径进行,速控步骤为式(3)Tafel反应。
图6中MoS2催化剂的塔菲尔斜率为150mV/dec,H+在配位不饱和金属原子上吸附活化生成中间物种M-H*(Volmer反应)是速控步骤。N-MoS2催化剂的塔菲尔斜率为60~89mV/dec,M-H*的电化学脱附(Heyrovsky 反应)为速控步骤。以上结果说明在MoS2和M-MoS2表面通过Volmer-Heyrovsky 路径析氢,且N 的掺杂降低了MoS2的塔菲尔斜率,速控步骤从Volmer 反应变为Heyrovsky 反应,提高了析氢反应的活性。这是由于相比MoS2催化剂,N-MoS2中存在较多具有催化活性的不饱和配位Mo原子,足够的H+可以在MoS2催化剂上吸附活化,Volmer 反应不再是析氢反应的速控步骤。但由于Mo-H 相互作用较强,H*在MoS2表面无法快速迁移,Mo-H*难以通过复合脱附(Tafel 反应)快速析氢,因而仍需要电化学驱动通过Heyrovsky 反应析氢。图6 显示,不同N 掺杂量的N-MoS2催化剂的塔菲尔斜率不同,说明不同N-MoS2催化剂上的析氢反应速率不同。反应速率的差异不仅与不饱和配位点的暴露量有关,还可能与Heyrovsky 反应中活性位上H*的反应脱附能力有关。
有研究发现,氢电极析氢反应活性与“金属-氢”(M-H)相互作用强度有关。根据Trasatti[40]总结的交换电流密度与M-H键能之间的火山关系图,在适中的M-H 相互作用强度时,析氢反应速率最快,交换电流密度达到最大值。而Pt 作为目前最好的电催化析氢催化剂,Pt-H*相互作用强度适中,H*在Pt 表面可以快速迁移[41],且表面的H 覆盖度较高,Pt-H*可以通过Tafel反应快速析氢。其塔菲尔斜率为30mV/dec,电催化析氢的反应路径为Volmer-Tafel路径[37,39]。
本研究中,虽然N 掺杂增加了MoS2中配位不饱和Mo原子的暴露量,提高了MoS2表面的H覆盖度,但是由于Mo-H相互作用较强,H在MoS2表面无法快速迁移,Mo-H*难以通过Tafel 反应快速析氢,只能通过Heyrovsky 反应析氢。因此,对于高HER 活性的MoS2催化剂的设计,不仅要提高活性位点的暴露量,还要优化活性位点与H之间的相互作用强度,使析氢反应通过Volmer-Tafel 路径高效进行。在本工作中,N 掺杂MoS2催化剂中Mo 原子电子密度较高,富电子的Mo 会削弱Mo-H*,有利于吸附在MoS2上的H*通过Heyrovsky 反应与H+结合生成H2[37]。其中N-MoS2-0.1催化剂中Mo 3d的结合能相对于MoS2负移最多,电子密度最高,对Mo-H*的削弱程度大,N-MoS2-0.1 催化剂的塔菲尔斜率最小,析氢活性最高。N-MoS2催化剂HER催化活性的提高可归结为N掺杂使得催化剂上配位不饱和Mo 原子的增多和富电子的Mo 对Mo-H*的弱化。因此,高效电催化析氢催化剂的设计不仅需要增加活性位点的暴露,还需要调控“金属-氢”的相互作用强度,使催化剂的HER 活性向Pt 靠近并取代Pt。
如图7 所示,经过1000 次循环后得到的极化曲线与初始极化曲线几乎重叠。在电流密度为0~20mA/cm2范围内,过电位几乎无变化,表明N-MoS2-0.1催化剂具有良好的电催化析氢稳定性。推测在电催化析氢反应中N 原子不会被排出MoS2晶格,N原子掺杂生成的活性位点不会发生变化。

图7 N-MoS2-0.1催化剂的循环稳定性测试
3 结论
以钼酸钠为钼源、L-半胱氨酸为硫源、双氰胺为氮源,通过一步水热法成功合成了N 掺杂的MoS2催化剂。此外,通过调变氮源与钼源的原子比得到了不同N 掺杂量的N-MoS2催化剂。表征结果显示N 均匀掺进了MoS2晶格,且N 掺杂使得Mo原子的电子密度增加,并生成大量不饱和配位点。与MoS2相比,N-MoS2催化剂的塔菲尔斜率较低,析氢反应(Volmer-Heyrovsky 路径)的速控步骤由Volmer 反应变为Heyrovsky 反应,表现出显著提高的电催化析氢性能。其中N/Mo 原子比为0.1 的NMoS2-0.1催化剂表现出了最高的电催化析氢活性和较好的稳定性。HER 催化活性的提高可归结为NMoS2催化剂上不饱和配位点的增多和富电子的Mo对Mo-H*的弱化。作为稳定高效的电催化析氢催化剂,N掺杂MoS2纳米催化剂在电解水制氢中具有广阔的应用前景。
