高通量测序文库质量控制技术研究进展
2024-02-22崔梦楠郭彦武雅蓉裴广倩崔玉军
崔梦楠,郭彦,武雅蓉,裴广倩,崔玉军
综 述
高通量测序文库质量控制技术研究进展
崔梦楠,郭彦,武雅蓉,裴广倩,崔玉军
军事科学院军事医学研究院微生物流行病研究所,北京 100071
作为生命科学和医学领域中的关键性支撑技术,高通量测序已得到快速发展并日趋成熟。该技术工作流程可分为核酸提取、文库构建、上机测序、数据分析等,其中文库构建是承上启下的关键步骤,文库构建的效果受制于上游样品质量,同时会对测序数据产出后的数据分析造成影响。对文库构建质量控制技术的选择和实施是提高结果可靠性、降低测序数据误差的重要保证。本文对文库构建质量控制技术进行深入综述,总结评价其原理、优缺点、适用范围,并对实际应用场景中相关技术的选择进行了论述,以期为科研人员、疾病预防控制人员等在选择文库质量控制技术时提供理论依据与参考,从而促进高通量测序工作的质量和效率。
高通量测序文库;文库浓度;文库大小分布;质量控制;技术评价
高通量测序又称下一代测序(next generation sequencing),相较于Sanger测序,通量高,运行一次能读取几十万到几百万条DNA分子的序列,在鉴定病原、揭示病原微生物变异与进化、微生物群落组成等方面具有独特的优势[1~3],已被广泛应用在生命科学和医学领域[4]。由于高通量测序仪无法识别天然的DNA分子,所以测序前需进行文库的构建,即在DNA分子上连接接头序列,使之能够被测序仪捕获,进行克隆扩增反应以激发信号[5]。例如,Illumina测序采取了边合成边测序,通过桥式PCR形成簇,将文库序列与测序芯片(flow cell)连接固定。
文库构建的实验流程虽然较为繁琐复杂,但意义重大。文库的质量与样品的质量具有一定的相关性,其在一定程度上可识别质量较差或浓度不足的样品,低质量的样品会影响文库的转化率、测序的深度、复杂性和均一性,同时对质量高的文库进行上机测序也是测序成功的前提。对质量不合格的文库进行上机测序会降低数据的产出,达不到有效数据量进而影响下游分析,甚至会导致测序失败,无法充分发挥测序平台的功能,浪费样品、昂贵的试剂、用户时间和仪器[6]。此外一张芯片的理论数据量一般远远大于单个样品产生的实际测序数据量,在实际操作过程中,往往对多样品文库进行混合(pooling)[7],以此来提高经济效益、时间效益和通量。当进行上机测序的文库浓度较低时,产生的有效数据量较少,增大测序成本、延长测序周期;当文库浓度较高时,影响上机测序时的捕获效率,严重时会导致测序异常;当部分文库浓度较高、部分文库浓度较低时,会造成样品的测序深度不同,较高的测序深度会加重数据处理负担,提高测序成本;较低测序深度可能会导致一些基因组区域的覆盖不足,影响特定区域的检测能力,结果可信度较差。因此,对文库的质量控制是实现最优数据产量、提高实验室效率和测序通量的重要保障。
文库的质量控制技术通常包括对文库浓度的评价和大小分布的评价,常用的浓度测定技术有紫外吸收技术[8~10]、荧光染料技术[11~13]、实时荧光定量PCR(quantitative real-time PCR, qPCR)[14]技术和微滴数字PCR(droplet digital PCR,ddPCR)技术[15]等。常用的大小分布测定技术有琼脂糖凝胶电泳技术[16]和微流控芯片技术[17]等。目前国内外领域对这些质量控制技术缺乏系统性的评价,本文从各技术的原理、优缺点、适用范围等进行分析,同时也对常见技术进行横向对比,总结了不同技术的应用场景,并展望了文库质量控制技术的发展,以期为文库的评价、文库混合等提供可靠的选择依据,为获得高质量的测序数据奠定基础,进而提高测序通量、缩短测序周期、降低测序成本。
1 文库浓度的质量控制技术
1.1 紫外吸收技术
紫外吸收技术是利用核酸的紫外吸收特性进行浓度定量,是较常用的技术,常用的仪器有美国赛默飞公司的NanoDrop。NanoDrop在测定前不需要进行额外的操作,是一种简单、快速的测定文库质量技术,但是灵敏度较低(1ng/μL)。Hussing等[18]用NanoDrop测定扩增子文库的浓度,发现浓度被高估,原因可能是双链DNA、单链DNA、游离核苷酸等在260nm处均有光吸收信号,敏感性较差。当文库浓度越低时,NanoDrop的测定值越不稳定,波动越大。Harris等[19]对一个文库通过NanoDrop进行三次重复测定,评价其精确度,结果表明,变异系数较高,为12%,变异系数越高,表明均一化程度越低。Hosomichi等[20]先以NanoDrop定量文库后再以等摩尔比例混合上机,发现文库的覆盖度变化较大。可见,紫外吸收测量法存在着一定的弊端,究其原因:一是对DNA、RNA、蛋白质没有选择性;二是其绝对值受其他污染物和碱基组成的影响较大;三是当DNA浓度较低时,精确性较差[21]。因此,NanoDrop适用于文库的初检或者作为不具有其他高灵敏度检测技术时的替代品。
1.2 荧光染料技术
荧光染料技术是通过测定染料与DNA文库结合时发出的荧光强度来定量DNA文库,荧光强度与DNA浓度成正比。荧光染料法操作便捷、快速、灵敏度较高、成本较低[22]。常用的定量DNA文库的荧光染料有Hoechst 33258和PicoGreen。Hoechst 33258染料能检测低至10pg/μL的DNA,PicoGreen染料能够定量低至25pg/μL的dsDNA。Qubit (美国赛默飞公司)是典型的通过荧光染料法定量DNA文库的仪器。与NanoDrop相比,Qubit最显著的特征是能提供更可靠的定量结果,准确率高。当定量10 ng/μL的DNA时,NanoDrop的测定错误率高达5%,而Qubit仅为1%[23]。当定量0.5 ng/μL的DNA时,采用Qubit进行10次重复测定,平均值为0.53ng/μL,95%置信区间为0.47~0.60 ng/μL[24]。当样品质量较差时,Qubit定量的成功率为80%[21]。
采用Qubit定量的DNA文库浓度高于基于电泳定量的浓度,与Qubit不能区分不同长度的DNA有关,使得引物二聚体、接头二聚体、未连接接头的gDNA、PCR产物等被计算在内[18]。有研究表明,当使用PicoGreen染料时,定量结果受DNA片段大小分布的影响,浓度随片段的增加而降低[25]。当长度小于23kb时,浓度被低估[26]。Qubit定量是一个较便宜、较精确的定量平台,操作流程快速简便,但是每次只能对一个样品进行检测,当样品数量较多时,耗费时间较长。
1.3 qPCR技术
qPCR技术是指在PCR反应体系中加入荧光基团,通过监测荧光信号的变化来定量浓度。由于其特异性引物的存在,qPCR技术能够估计出文库中可扩增的目标片段的数量,具有较高的敏感性和特异性,能够准确排除单端或双端都不连接接头的不可测序文库的干扰,在准确定量在5′和3′端连接接头的分子片段时具有明显优势。目前,根据荧光标记的方法不同,qPCR分析检测技术分为基于荧光探针法(TaqMan)[27]和荧光染料法(SYBR-Green)[28]。由于SYBR-Green 法不需要设计荧光探针,方法更为灵活且便宜[29]。Dang等[30]研究表明,TaqMan法和SYBR法在定量文库浓度上具有相似性。通过qPCR技术定量的文库浓度的好坏与利用测序后得到的数据进行下游分析后的得到的总序列数多少具有相关性,这可能是因为后续的桥式PCR和乳浊液PCR也是PCR反应[31,32]。
PCR反应容易受多种因素的影响,是一个较敏感的反应。增加PCR反应会导致文库序列异源双链的减少,从而使序列的原始比率失真,影响测序质量[33],并损失珍贵的样品[34]。当扩增过短的片段,高GC:AT含量和DNA/Taq聚合酶保真度较低时,序列的异质性可能会随着PCR的扩增反应而改变[35]。qPCR文库定量技术不是绝对定量方法,不能区分接头二聚体,不适合接头二聚体浓度高的片段,依赖于生物分析仪对文库片段大小分布的检测[28]。而在复杂的文库中,引物和污染物会影响片段大小测定的准确性,最终使得加载到测序芯片上的文库的量有一定的波动。但与紫外吸收技术和荧光染料技术相比,虽然qPCR技术成本较高(为6~12倍),花费时间较长(为5~10倍)[18]。但是当对文库质量要求较高时,还应采用qPCR技术进行定量,因为与测序试剂、重测序成本相比,qPCR技术与其他两种定量技术在人工操作时间和价格的差距可忽略不计。
1.4 ddPCR技术
1999年,Volgelstein 等[36]提出了数字PCR (digital PCR,dPCR)。dPCR最开始主要有以下两种模式:一是使用微孔或微流控室将反应分成许多纳升级反应室,分别进行PCR扩增,检测阳性液滴数量,根据泊松分布进行绝对定量。虽然微流控芯片简化了反应程序,但是难以实现大通量及规模化;二是利用BEAMing,该模式基于乳液PCR,即增强型dPCR。原理是在磁珠上进行dPCR后,再利用荧光杂交探针标记,最终被传统的流式细胞读取,该技术虽然通量高,但工作的复杂程度也较高。2011年,Hindson等[37]开发出了油包水液滴的ddPCR,即使用微流体和特定表面活性剂化学物质,将PCR样品分成油包水液滴进行dPCR,原理如图1所示。在ddPCR中,一个样本被分割成几十至几万份微滴至不同的反应单元,每个单元至少包含一个拷贝的目标分子(DNA模板),目标分子能在不同的反应单元里进行PCR扩增,扩增反应结束后通过收集各个反应单元的荧光信号,进行统计学分析。ddPCR的操作流程和使用试剂与TaqMan探针法类似。
ddPCR可以根据簇的荧光强度的不同来判定文库片段的质量。基于ddPCR的两端水解探针,当文库两端连接P5、P7时,呈双阳现象;当只有接头二聚体时,PCR扩增效率最高,荧光最强,能够明显地与前者区分开。PCR扩增效率与接头二聚体的长度呈反比。当文库的质量较好时,往往沿着双正簇分布在FAM/HEX通道2D散点图的右上方,无其他FAM荧光。Heredia等[15]对通过ddPCR定量后的12个文库按等摩尔比例进行上机测序,发现每个文库对应的序列数无显著差别,该结果进一步体现了ddPCR较强的质量控制能力。ddPCR还能提供文库构建过程中的动态信息,如接头连接、PCR扩增等。Aigrain等[37]用ddPCR技术从每个文库构建步骤后剩余的DNA含量、片段末端连接接头的比例、扩增后带有P5/P7的比例三个方面评价了不同文库构建方法,发现接头连接效率是文库构建过程中的关键步骤,接头连接效率较低会影响文库的复杂度。不同的文库构建方法中,连接和PCR产量呈相反变化趋势,这可能是因为起始DNA投入量低或者连接效率低,进行PCR反应的带有DNA片段的接头的量较少,也可能是因为连接效率较高或DNA投入量较大,大量的DNA进行了PCR反应。较高的连接率能够保证样品的多样性,缩短所需的PCR循环数,从而避免因PCR过程带来的偏差[38]。
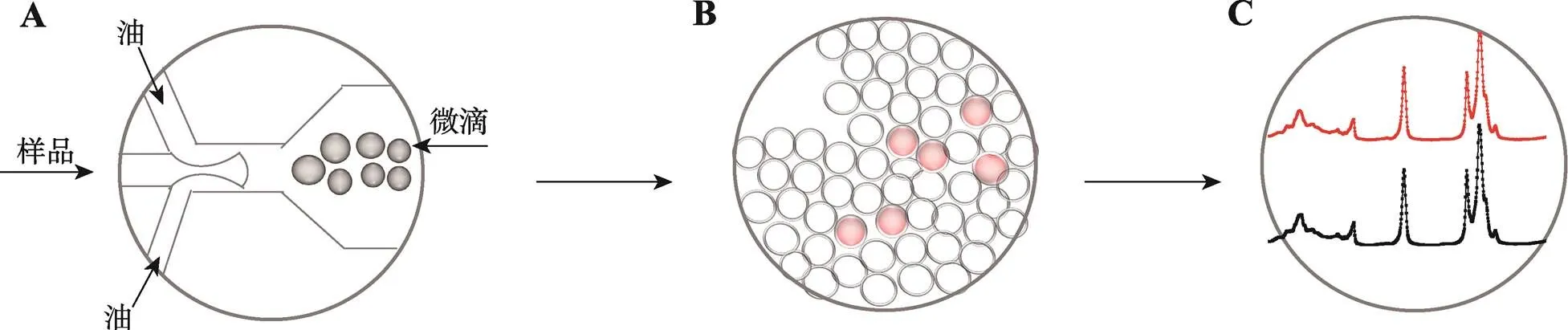
图1 ddPCR的原理
A:微滴制备。配制含样品、PCR酶、dNTP、缓冲液、特定引物、Taqman探针的反应体系,并将反应体系置于微滴发生器中,使一样本被分割成几十至几万份微滴至不同的反应单元,每个反应单元至少包含一个拷贝的目标分子(DNA模板)。B:微滴PCR扩增。在每个反应单元中单独进行PCR反应,只有含有目标分子的微滴在PCR扩增后显示出高荧光。C:微滴读取和数据分析。读取每个微滴荧光值,确定阴性、阳性微滴数,并通过泊松统计分析计算原始样品中目标DNA模板浓度。
不同文库浓度的质量控制技术具有不同的优劣势(表1)。与前面的评价技术相比,ddPCR是绝对定量,不依赖于具有特定大小分布的标准品,不依赖于校准物的扩增效率,避免了qPCR中真实样品扩增效率可能与校准物扩增效率不同的问题,是一种准确度更高、置信度和可重复性更好的评价技术。
2 文库大小分布的质量控制技术
构建好的文库中可能会存在二聚体、小片段、大片段等非目的片段及片段峰过宽的问题,影响定量的准确度、精确度,从而干扰下游的生物信息学分析,因此需要对文库大小分布进行双重验证。常见的检测技术有传统的琼脂糖凝胶电泳、微流控芯片技术等。
2.1 琼脂糖凝胶电泳技术
琼脂糖凝胶电泳技术基于待分离样品中各种分子带电性质的差异以及分子本身的大小、形状的不同,从而在通过琼脂或琼脂糖作支持的凝胶介质时产生不同的迁移速度,来分离带电分子,是一种较经典的技术,目前仍常用于鉴定和纯化DNA片段。在电场作用下,DNA片段迁移距离与碱基对的对数成反比,可通过计算待测片段的移动距离与已知大小的标准品的移动距离的比例来定量未知片段的大小。琼脂糖凝胶电泳需要配胶、加染料、制胶板、上样/制样、电泳、检测等步骤,虽然原理、操作等较简单,成本较低,但是操作时间较长,分离精度较低。Chang等[16]利用高温高压制备了充分溶解的高浓度琼脂糖凝胶,发明了一种简单易操作的灌胶塑料盒灌制黏稠的HAG垂直凝胶,改进了适合小分子量核酸的缓冲液条件,可有效分离在10~100bp范围内相2bp或2nt的DNA片段,解决了琼脂糖凝胶浓度较低,低分子量核酸分辨率较低的问题,并且制备过程中不涉及危险化学试剂,安全、无毒。
该技术常联合采集设备来得到相应的凝胶电泳图像,对图像的精确处理和分析尤为重要。Ziraldo等[39]开发了基于ImageJ的插件,该插件可以用于分析连续型或离散型凝胶模式,在离散型模式下,基于条带的强度和宽度直接定量DNA分子的相对数量;在连续型模式下,通过从标准品衍生的叠加高斯电泳峰来预估DNA大小分布,是一种有效的且成本较低的分析文库大小分布的方式。与美国安捷伦公司TapeStation仪器的定量结果具有较高的一致性,且在峰较平缓时检测具有一定的优势,能够处理较低的信噪比电泳图。

表1 不同文库浓度评价技术的比较
a来源于官网说明,https://assets.thermofisher.com/TFS-Assets/CAD/manuals/nd-1000-v3.8-users-manual-8%205x11.pdf;b来源于官网说明,https://www.thermofisher.cn/document-connect/document-connect.html?url=https://assets.thermofisher.cn/TFS-Assets%2FLSG%2Fmanuals%2FMAN0017455_Qubit_1X_dsDNA_HS_Assay_Kit_UG.pdf;c来源于官网说明,https://www.bio-rad.com/sites/default/files/webroot/ web/pdf/lsr/literature/Bulletin_6407.pdf。
然而,由于传统的琼脂糖凝胶电泳分离精度较低、操作繁琐、自动化程度低,在分离鉴定大量重叠片段时存在着一定的困难,因此还需要高精度、高自动化、高效率的技术来解决该问题。
2.2 微流控芯片电泳技术
微流控芯片技术最早由Manz和Widmer[40]于20世纪90年代提出,他们的研究表明了在微通道网络中以电渗流为驱动力实施进样和电泳分离的可能性。随着科学技术的发展,在芯片上构造出电泳微流通道结构,包括进样系统、分离系统、检测系统等,当给予芯片一定的电压时,样品便在芯片上的显微蚀刻管道中进行毛细管电泳。在样品流动过程中,不同DNA片段根据其大小被分离。微流控芯片电泳技术是集分离、检测为一体的自动化、一体化、集成化的技术。
对芯片进行改进可以提高样品的通量,缩短时间,提高分选效率。Loughran等[41]对芯片进行修饰,构建了40条平行并列的阵列电泳通道,并用二甲基丙烯酰胺-烯丙基缩水甘油醚控制电渗流,在6 min内完成了10个DNA片段的分离检测。刘科辉等[42]通过改变介质和嵌入式荧光标记染料,实现了无胶筛分和激光诱导荧光检测,在75s内分离了12条DNA片段。Sun等[17]开发了一种新的荧光片段分析试剂,搭配美国珀金埃尔默公司商业化片段分析仪LabChip GX Touch使用,该检测技术整合了样品分离、检测、数据可视化和初步的数据分析报告,样品加载和数据获得过程是自动化的、快速化的,在96-孔板中最多可处理48个样品,每个样品处理时间为1 min。
目前,基于微流控芯片电泳技术的商业化平台有美国安捷伦公司的Bioanalyzer、TapeStation、Fragment Analyzer,美国珀金埃尔默公司的GX Touch等。他们主要由能够自动输送流体的注射泵、激发染料的LED芯片、监测荧光强度的变化的电荷耦合器件组成,无需进行凝胶制备、电泳、染色、脱色、成像等操作步骤,减少与有害物质的接触,所需进样量、试剂量等少,有利于保护珍贵样品,操作简单安全、数据可视化、分析自动化。此外,与传统的琼脂糖凝胶电泳技术相比,微流控芯片电泳技术准确度高、重复性好、灵敏度高。在500 bp片段的范围内,一般商业类仪器的分辨率有3~5bp的误差。Chiappetta等[43]通过计算ROC曲线得出Agilent 2100 Bioanalyzer (美国安捷伦公司)的灵敏度为86.7%,特异性为92.3%。当内标与测定片段长度差异较大时,计算偏差也较大,优化内标可进一步提高重复性。王洪霞等[44]设计了与测序片段相近的内标用于校正片段,发现可以纠正重复性较差的缺点,具有相同VNTR位点数的菌株的电泳峰完全重叠。
相较于传统的琼脂糖凝胶电泳技术,采用微流控芯片技术可以更好地、更精确地获得文库的大小及片段分布,且灵敏度更高,浓度低的片段也能被检测到,检测过程简单方便、高效,是一种化学品消耗少、分析时间短、分离效率高的文库大小分布定量技术,但是芯片价格较昂贵,当实验室经费紧张时,价格低廉的琼脂糖凝胶电泳技术更受到青睐(表2)。
3 文库质量控制技术的选择
不同的文库质量控制技术各有优缺点,在选择时,应根据实际需求和应用场景,对成本、准确度、精确度、时间、操作自动化、操作复杂度、投入量等进行综合考量。一般来说,成本和准确度、精确度、操作复杂度成正比,和操作自动化成反比。
当成本较宽松,但准确度要求较高时,如临床和法医DNA实验室,应选择qPCR分析技术对文库进行浓度质量控制。qPCR分析是一种灵敏度较高的定量技术,其定量值的大小与下游测序数据分析后的文库覆盖度具有较好的相关性;同时,该技术还能用于评估文库中扩增的目标分子的数量。虽然qPCR分析技术的成本较高,但与浓度质量控制技术间的价格差异相比,因质量控制不准确而导致的测序失败引起的测序试剂成本、重测序时间成本更高。因此,当对文库覆盖度的均一性要求较高,且不计成本时,应选择准确度、精确度较高的qPCR质量控制技术。虽然ddPCR能够对文库进行绝对定量,并且能够定量相对文库大小分布、文库构建过程中产生的接头二聚体的数量等,但因ddPCR操作较复杂,如微滴形成和PCR扩增、监测需在不同的仪器中完成,在实际应用中较少。
在需要压缩成本,且能接受文库覆盖度的差异性波动时,可以选择相对便宜、快速、简单的荧光染料分析技术,如Qubit。Qubit分析技术准确度较高,当用其测定已知浓度的双链DNA寡核苷酸时,浓度测定值与理论值较接近,且当浓度较低时(20~40pg/μL),也能被精确测定[45]。但是紫外吸收技术因其对DNA、RNA、蛋白质没有选择性,准确性、灵敏度都较差,在不具备其他高灵敏度检测装置时可用作文库质量控制的初检。当有其他技术选择时,不推荐用于文库浓度的测定。值得注意的是,Qubit分析技术不能区分不同长度的DNA,当文库中存在引物二聚体、接头二聚体、未连接接头的小片段、大片段时,会使文库的实际浓度被高估。
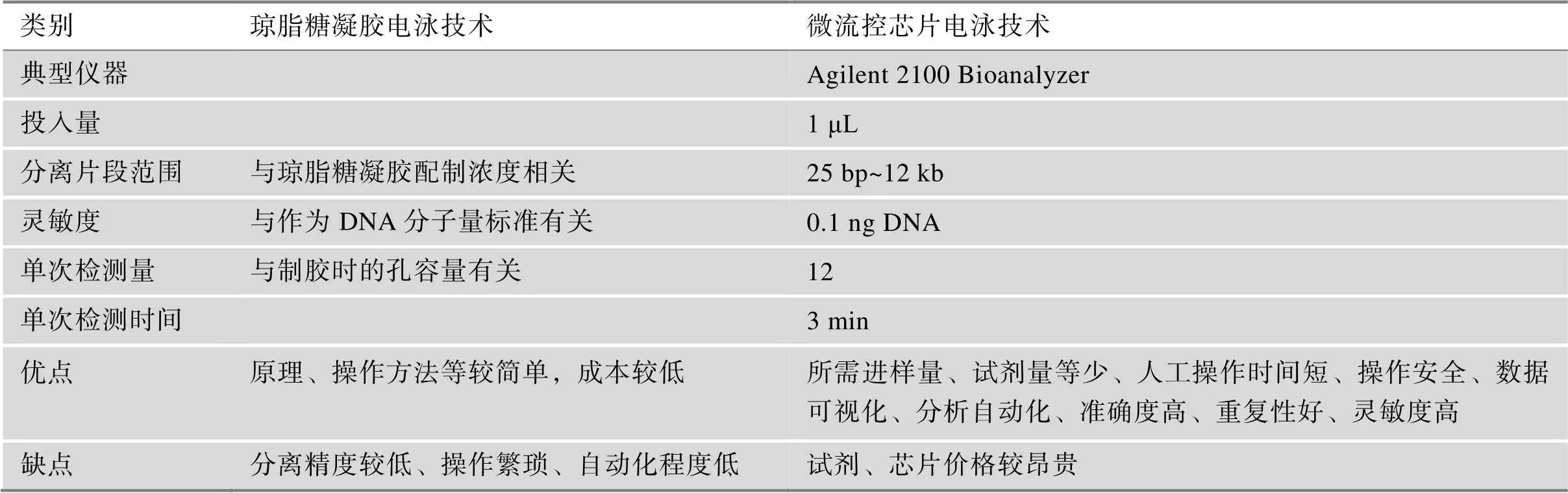
表2 不同文库大小分布评价技术的比较
文库浓度的质量控制技术通常需要和文库大小分布的质量控制技术协同应用,因为qPCR分析技术、Qubit分析技术均对不同长度的DNA没有选择性。在考虑成本因素时,可选择传统的琼脂糖凝胶电泳技术。不计成本时,可选择基于微流控芯片电泳技术的商用化平台,如美国安捷伦公司的Bioanalyzer、TapeStation、Fragment Analyzer、美国珀金埃尔默公司的GX Touch等。其中,Bioanalyzer和TapeStation操作简便,但是同一批次运行的样品量少,在这四种商业化仪器中,Bioanalyzer成本最高。Fragment Analyzer通量高,操作复杂,在无人值守的情况下,每24 h可分析上千个文库。GX Touch虽然能够提供可重复使用的芯片,但是清洗流程复杂,且在进行文库大小分布测定前需对文库浓度进行测定或估计,浓度过高会破坏芯片。此外,基于毛细管电泳技术的光鼎公司的Qsep也可实现对文库大小分布的测定,其所需投入量低,仅为0.5 ng/μL,分离片段范围可达15 bp~15 kb,运行一次可实现对96个文库的检测,单个文库检测时间大约为5 min。与基于微流控芯片电泳技术的商用化平台相比,成本较低。
极端特殊情况时,在保证规范操作和下游数据谨慎分析的前提下,可忽略文库的质量控制结果。如样本非常重要且难以或没时间重新获取时,即使文库构建结果不满足质量控制要求,也可尝试进行下一步测序操作。2019年底新冠肺炎疫情爆发时,Chen等[1]从两名感染异常肺炎的患者肺泡灌洗液样本中提取总RNA,通过Qubit对提取后的浓度进行测定,发现浓度低于0.5ng/μL(当加入1μL样品时,Qubit的检测下限),后采取靶向目标的Trio RNA-Seq试剂盒(瑞士帝肯公司)构建文库,应用Illumina Miseq平台进行双端测序。经生物信息学分析,鉴定出一种新型的冠状病毒,为疫情防控奠定了基础。
4 结语与展望
高通量测序技术已经从专业实验室走向常规分子生物学实验室和检测诊断实验室。随着其应用范围的不断扩展,整个测序流程的质量控制变得日益关键。文库构建作为高通量测序中的核心环节之一,其质量控制对于确保获得准确和有用的测序数据至关重要。目前高通量测序文库质量控制技术可选择性较多,但各自存在局限性,例如,一些准确度较高的技术操作复杂,难以自动化;而成本低、操作简便的技术,则可能在文库浓度和片段大小的准确测定方面有所不足。因此,应根据不同的实验条件和目的,灵活选择合适的文库构建质量控制技术。
随着生物技术的不断进步,特别是高通量测序平台技术的高速发展,我们有理由相信文库构建质量控制技术将持续得到改进和完善,未来的技术发展将趋向智能化、简便化和低成本化,而这将进一步简化操作流程,降低对专业人员和实验室条件的依赖。这些进步将使高通量测序技术在生命科学研究、诊断与个性化医疗、公共卫生监测等领域发挥更大作用,为社会带来更加显著的应用价值。
[1] Chen LJ, Liu WY, Zhang Q, Xu K, Ye GM, Wu WC, Sun ZY, Liu F, Wu KL, Zhong B, Mei Y, Zhang WX, Chen Y, Li YR, Shi M, Lan K, Liu YL. RNA based mNGS approach identifies a novel human coronavirus from two individual pneumonia cases in 2019 Wuhan outbreak., 2020, 9(1): 313–319.
[2] Zheng HY, Yan L, Yang C, Wu YR, Qin JL, Hao TY, Yang DJ, Guo YC, Pei XY, Zhao TY, Cui YJ. Population genomics study of Vibrio alginolyticus., 2021, 43(4): 350–361.郑宏源, 闫琳, 杨超, 武雅蓉, 秦婧靓, 郝彤宇, 杨大进, 郭云昌, 裴晓燕, 赵彤言, 崔玉军. 溶藻弧菌群体基因组学研究. 遗传, 2021, 43(4): 350–361.
[3] Wang GZ, Long J, Zhuang Y, Leng X, Zhang YQ, Liu LBX, Fu JW, Chen Y, Li CQ, Zhou Y, Huang B, Feng CC. Application of metagenomic next-generation sequencing in the detection of pathogens in spinal infections., 2023, 23(6): 859–867.
[4] Børsting C, Morling N. Next generation sequencing and its applications in forensic genetics., 2015, 18: 78–89.
[5] Liu YL, Xu C, Sun YZ, Chen X, Dong WP, Yang XY, Zhou SL. Method for quick DNA barcode reference library construction., 2021, 11(17): 11627–11638.
[6] Laurie MT, Bertout JA, Taylor SD, Burton JN, Shendure JA, Bielas JH. Simultaneous digital quantification and fluorescence-based size characterization of massively parallel sequencing libraries., 2013, 55(2): 61–67.
[7] Modi A, Vai S, Caramelli D, Lari M. The Illumina sequencing protocol and the NovaSeq 6000 system., 2021, 2242: 15–42.
[8] Glasel JA. Validity of nucleic acid purities monitored by 260nm/280nm absorbance ratios., 1995, 18(1): 62–63.
[9] Huberman JA. Importance of measuring nucleic acid absorbance at 240 nm as well as at 260 and 280 nm., 1995, 18(4): 636.
[10] Manchester KL. Value of A260/A280 ratios for measurement of purity of nucleic acids., 1995, 19(2): 208–210.
[11] Singer VL, Jones LJ, Yue ST, Haugland RP. Characterization of picoGreen reagent and development of a fluorescence-based solution assay for double-stranded dna quantitation., 1997, 249(2): 228–238.
[12] Le Pecq JB, Paoletti C. A new fluorometric method for RNA and DNA determination., 1966, 17(1): 100–107.
[13] Kapuscinski J. DAPI: a DMA-specific fluorescent probe., 1995, 70(5): 220–233.
[14] Heydt C, Fassunke J, Künstlinger H, Ihle MA, König K, Heukamp LC, Schildhaus HU, Odenthal M, Büttner R, Merkelbach-Bruse S. Comparison of pre-analytical FFPE sample preparation methods and their impact on massively parallel sequencing in routine diagnostics., 2014, 9(8): e104566.
[15] Heredia NJ. Droplet Digital™ PCR next-generation sequencing library qc assay., 2018, 1768: 477–488.
[16] Chang LL, Wang D, Peng CZ, Wang Q, Xu BQ, Tong Z. A method for high-concentration agarose gel preparation and its application in high-resolution separation of low- molecular-weight nucleic acids and proteins., 2023, 231: 123358.
[17] Sun YL, Lu ZX, Miller M, Perroud T, Tong YH. Application of microfluidic chip electrophoresis for high-throughput nucleic acid fluorescence fragment analysis assays., 2023, 5(1): lqad011.
[18] Hussing C, Kampmann ML, Mogensen HS, Børsting C, Morling N. Quantification of massively parallel sequencing libraries -a comparative study of eight methods., 2018, 8(1): 1110.
[19] Harris JK, Sahl JW, Castoe TA, Wagner BD, Pollock DD, Spear JR. Comparison of normalization methods for construction of large, multiplex amplicon pools for next-generation sequencing., 2010, 76(12): 3863–3868.
[20] Hosomichi K, Mitsunaga S, Nagasaki H, Inoue I. A bead-based normalization for uniform sequencing depth (BeNUS) protocol for multi-samples sequencing exemplified by HLA-B., 2014, 15(1): 645.
[21] Masago K, Fujita S, Oya Y, Takahashi Y, Matsushita H, Sasaki E, Kuroda H. Comparison between fluorimetry (qubit) and spectrophotometry (nanodrop) in the quantification of DNA and RNA extracted from frozen and FFPE tissues from lung cancer patients: a real-world use of genomic tests., 2021, 57(12): 1375.
[22] Tuononen K, Mäki-Nevala S, Sarhadi VK, Wirtanen A, Rönty M, Salmenkivi K, Andrews JM, Telaranta-Keerie AI, Hannula S, Lagström S, Ellonen P, Knuuttila A, Knuutila S. Comparison of targeted next-generation sequencing (NGS) and real-time PCR in the detection of EGFR, KRAS, and BRAF mutations on formalin-fixed, paraffin-embedded tumor material of non-small cell lung carcinoma-superiority of NGS., 2013, 52(5): 503–511.
[23] Sah S, Chen LJ, Houghton J, Kemppainen J, Marko AC, Zeigler R, Latham GJ. Functional DNA quantification guides accurate next-generation sequencing mutation detection in formalin-fixed, paraffin-embedded tumor biopsies., 2013, 5(8): 77.
[24] Simbolo M, Gottardi M, Corbo V, Fassan M, Mafficini A, Malpeli G, Lawlor RT, Scarpa A. DNA qualification workflow for next generation sequencing of histopathological samples., 2013, 8(6): e62692.
[25] Holden MJ, Haynes RJ, Rabb SA, Satija N, Yang K, Blasic JR. Factors affecting quantification of total DNA by UV spectroscopy and PicoGreen fluorescence., 2009, 57(16): 7221–7226.
[26] Georgiou CD, Papapostolou I. Assay for the quantification of intact/fragmented genomic DNA., 2006, 358(2): 247–256.
[27] Navarro E, Serrano-Heras G, Castaño MJ, Solera J. Real-time PCR detection chemistry., 2015, 439: 231–250.
[28] Robin JD, Ludlow AT, LaRanger R, Wright WE, Shay JW. Comparison of DNA quantification methods for next generation sequencing., 2016, 6: 24067.
[29] Arya M, Shergill IS, Williamson M, Gommersall L, Arya N, Patel HRH. Basic principles of real-time quantitative PCR., 2005, 5(2): 209–219.
[30] Dang J, Mendez P, Lee S, Kim JW, Yoon JH, Kim TW, Sailey CJ, Jablons DM, Kim IJ. Development of a robust DNA quality and quantity assessment qPCR assay for targeted next-generation sequencing library preparation., 2016, 49(4): 1755–1765.
[31] Dressman D, Yan H, Traverso G, Kinzler KW, Vogelstein B. Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations., 2003, 100(15): 8817–8822.
[32] Fedurco M, Romieu A, Williams S, Lawrence I, Turcatti G. BTA, a novel reagent for DNA attachment on glass and efficient generation of solid-phase amplified DNA colonies., 2006, 34(3): e22.
[33] Parkinson NJ, Maslau S, Ferneyhough B, Zhang G, Gregory L, Buck D, Ragoussis J, Ponting CP, Fischer MD. Preparation of high-quality next-generation sequencing libraries from picogram quantities of target DNA., 2012, 22(1): 125–133.
[34] Li MK, Stoneking M. A new approach for detecting low-level mutations in next-generation sequence data., 2012, 13(5): R34.
[35] Mamedov TG, Pienaar E, Whitney SE, TerMaat JR, Carvill G, Goliath R, Subramanian A, Viljoen HJ. A fundamental study of the PCR amplification of GC-rich DNA templates., 2008, 32(6): 452–457.
[36] Vogelstein B, Kinzler KW. Digital PCR., 1999, 96(16): 9236–9241.
[37] Aigrain L, Gu Y, Quail MA. Quantitation of next generation sequencing library preparation protocol efficiencies using droplet digital PCR assays-a systematic comparison of DNA library preparation kits for Illumina sequencing., 2016, 17: 458.
[38] Seguin-Orlando A, Schubert M, Clary J, Stagegaard J, Alberdi MT, Prado JL, Prieto A, Willerslev E, Orlando L. Ligation bias in Illumina next-generation DNA libraries: implications for sequencing ancient genomes., 2013, 8(10): e78575.
[39] Ziraldo R, Shoura MJ, Fire AZ, Levene SD. Deconvolution of nucleic-acid length distributions: a gel electrophoresis analysis tool and applications., 2019, 47(16): e92.
[40] Manz A, Graber N, Widmer HM. Miniaturized total chemical analysis systems: a novel concept for chemical sensing., 1990, 1(1–6): 244–248.
[41] Loughran M, Cretich M, Chiari M, Suzuki H. Separation of DNA in a versatile microchip., 2005, 107(2): 975–979.
[42] Liu KH, Liang N, Yao B, Luo GA, Wang YM. Development of Laser-induced fluorescence detector for deoxyribonucleic acid fragments seperation by microfluidic chip., 2005, 33(9): 1350–1353.刘科辉, 梁宁, 姚波, 罗国安, 王义明. 微流控芯片-激光诱导荧光检测器的研制及核酸片段分离检测中应用. 分析化学, 2005, 33(9): 1350–1353.
[43] Chiappetta C, Anile M, Leopizzi M, Venuta F, Della Rocca C. Use of a new generation of capillary electrophoresis to quantify circulating free DNA in non-small cell lung cancer., 2013, 425: 93–96.
[44] Wang HX, Cui ZG, Xiong LF, Zhang LJ, Kan B. Study on multiple locus VNTRs analysis ofby nucleic acid separation technology based on microfluidics., 2009, 24(3): 209–212.王洪霞, 崔志刚, 熊礼凤, 章丽娟, 阚飙. 基于微流控的核酸片段分离技术用于伤寒沙门菌MLVA分型的研究. 疾病监测, 2009, 24(3): 209–212.
[45] Hussing C, Kampmann ML, Mogensen HS, Børsting C, Morling N. Comparison of techniques for quantification of next-generation sequencing libraries., 2015, 5: e276–e278.
Progress on the quality control technology of next generation sequencing library
Mengnan Cui, Yan Guo, Yarong Wu, Guangqian Pei, Yujun Cui
100071,
As a key supporting technology in the fields of life sciences and medicine, high-throughput sequencing has developed rapidly and become increasingly mature. The workflow of this technology can be divided into nucleic acid extraction, library construction, sequencing, and data analysis. Among these, library construction is a pivotal step that bridges the previous and subsequent stages. The effectiveness of library construction is contingent on the quality of upstream samples and also impacts the data analysis following sequence data output. The selection and implementation of library construction quality control techniques are crucial for enhancing the reliability of results and reducing errors in sequencing data. This review provides an in-depth discussion of library construction quality control techniques, summarizing and evaluating their principles, advantages and disadvantages, and applicability. It also discusses the selection of relevant technologies in practical application scenarios. The aim is to offer theoretical foundations and references for researchers, disease prevention and control personnel, and others when choosing library quality control techniques, thereby promoting the quality and efficiency of high-throughput sequencing work.
next generation sequencing library; library concentration; size distribution of the library; quality control; technology analysis
2023-10-23;
2023-12-22;
2024-01-05
崔梦楠,硕士,实验师,高通量测序和数据解读。E-mail:mengncui@163.com
崔玉军,博士,研究员,基因组流行病学。E-mail:cuiyujun.new@gmail.com
10.16288/j.yczz.23-262
(责任编委: 刘钢)
