木薯花叶病毒AC4蛋白与AtPARN互作研究
2024-02-21刘琳玉赵平娟符艳刘志昕任艳利张秀春
刘琳玉 赵平娟 符艳 刘志昕 任艳利 张秀春


关键词:斯里兰卡木薯花叶病毒(SLCMV);AC4蛋白;聚腺苷酸特异性核糖核酸酶(PARN);酵母双杂交(Y2H);荧光双分子互补(BiFC)
木薯花叶病毒(Cassavamosaicgeminivirus,CMVs)引发的木薯花叶病(cassavamosaicdisease,CMD)严重危害木薯产业发展和粮食安全[1]。2018年在我国海南、福建首次报道CMD,随后在其他种植区也有报道[2]。斯里兰卡木薯花叶病毒(SriLankancassavamosaicvirus,SLCMV)是首先在斯里兰卡发现的引发木薯花叶病的一个株系,在分类上属于双生病毒科(Geminiviridae)菜豆金黄花叶病毒属(Begomovirus)[3-4]。SLCMV是典型的双组分双生病毒,其基因组由DNA-A和DNA-B两个环状组分组成。DNA-A组分正义链上编码AV1(CP)和AV2两个蛋白,反义链编码4个蛋白,分别为AC1、AC2、AC3和AC4;DNA-B组分则编码BV1和BC1[3,5]。据报道,SLCMVAC4蛋白可抑制寄主PTGS提高病毒致病性,是病毒致病因子[6]。
无义介导的mRNA降解(nonsense-mediatedmRNAdecay,NMD)是真核细胞mRNA降解的主要途径之一,通过识别由于出现提前终止密码子(prematureterminationcodons,PTCs)而产生的在3′端有长非编码区域的mRNA(也称为longnon-codingRNAs,lncRNAs),并利用核糖核酸酶迅速进行降解[7-8]。越来越多的研究显示,NMD不仅是真核生物重要的mRNA数量、质量调控机制,还与转录后基因沉默(posttranscriptionalgenesilencing,PTGS)同样能降解病毒RNA,是真核生物普遍存在的抗病毒防御机制。NMD途径降解RNA包括PTCs识别、脱腺苷酸、脱帽和最后的核酸外切酶降解4个连续过程,UPF1(up-frameshiftprotein1)、PARN[poly(A)-specificribonuclease]、DCP2(decapping2)和XRN4(exoribonuclease4)分别是上述4个过程中的关键蛋白[9-11]。在真核生物中,去腺苷酸化是mRNA降解的首要限速步骤。PARN不仅在降解mRNApoly(A)尾中发挥關键作用,还参与非编码RNA的加工成熟、DNA损伤以及肿瘤等疾病发生过程[12-13]。病毒是专性寄生生物,必须逃避或耐受寄主细胞的降解才能成功感染寄主[14]。病毒如何调控NMD的抗病毒防御功能的分子机制研究已取得一些进展。有研究发现,系统沉默upf1后摩擦接种烟草花叶病毒(Tobaccomosaicvirus,TMV),病毒RNAs与蛋白的表达量均有增加,结果表明TMV的致病性可能与upf1有关[15]。萝卜花叶病毒(Turnipmosaicvirus,TuMV)中2个沉默抑制子VPg、HC-Pro分别与DCP2、XRN4相互作用并降低NMD的抗病毒防御功能,增强致病性[16]。GARCIA等[17]发现NMD能够靶标马铃薯X病毒(PotatovirusX,PVX)并限制其复制,研究结果显示PVX中2个含长的3′UTR亚基因组均会被NMD降解,并且在烟草中过量表达upf1能够抑制另一正链RNA病毒——芜菁皱缩病毒(Turnipcrinklevirus,TCV)的复制,说明抑制RNA病毒复制可能是NMD的普遍功能。
SLCMV复制过程中产生大量的lncRNAs,这些lncRNAs是如何逃避或耐受被寄主NMD降解,目前还知之甚少[18]。尚不明确SLCMVAC4蛋白是否可与NMD信号通路中的关键蛋白相互作用而抑制寄主NMD功能从而提高病毒致病性。本研究利用酵母双杂交(yeasttwo-hybrid,Y2H)技术和荧光双分子互补(BiFC)试验,对SLCMVAC4蛋白与拟南芥(Arabidopsisthaliana)NMD信号通路中的关键蛋白PARN之间的相互作用进行研究,研究结果将为阐明木薯花叶病毒是如何调控NMD的抗病毒防御功能的分子机理奠定基础。
1材料与方法
1.1材料
本氏烟草(Nicotianabenthamiana)种植于植物生长培养房,光照16h,黑暗8h,培养温度为20℃。酵母表达载体pGADT7、pGBKT7、pGBKT7-PARN以及BiFC载体:pGN1和pPARN-P28均为本实验室保存;pGADT7-T与pGBKT7-53(阳性对照)、pGADT7-T与pGBKT7-Lam(阴性对照)共转化的酵母菌、沉默抑制子表达载体pPTN-P19均为本实验室保存。
质粒小量中提取试剂盒和DNAMarker均购自天根生化科技(北京)有限公司;限制性内切酶BamHⅠ、EcoRⅠ等购自宝日医生物技术(北京)有限公司(TaKaRa);高效无缝克隆试剂盒购自莫纳生物科技有限公司;琼脂糖凝胶DNA试剂盒购自上海易汇生物科技有限公司(OMEGA);大肠杆菌(Escherichiacoli)DH5α、GV3101感受态细胞和酵母AH109菌株感受态细胞均购自上海唯地生物技术有限公司;SD/-Trp/-Leu、SD/-Trp/-Leu/-His、SD/-Trp/-Leu/-His/-Ade培养基均购自北京泛基诺科技有限公司;LB培养基购自生工生物工程(上海)股份有限公司;基因合成和测序由华大基因科技有限公司完成。
1.2方法
1.2.1酵母表达载体pGADT7-AC4的构建参考已报道的SLACMV序列(GenBank:KT861468.1),人工合成两端分别添加酵母猎物载体pGADT7限制性核酸内切酶EcoRⅠ和BamHⅠ位点两侧序列的目的片段Y2H-AC4(表1)。利用无缝克隆试剂盒将目的片段与经限制性核酸内切酶EcoRⅠ和BamHⅠ双酶切后的酵母猎物载体pGADT7进行连接反应,连接产物转入大肠杆菌菌株DH5α感受态细胞中,将其涂布于含氨苄青霉素(50mg/L)的LB平板上,置于恒温培养箱中,于37℃,倒置培养12h。然后,挑取3~5个单菌落,使用通用引物pGADT7-F和pGADT7-R(表2)对其进行PCR鉴定,PCR反应体系(20μL):各0.4μL引物(10μmol/L),2μL10×EasyTaqPCRBuffer(含Mg2+),1.5μLdNTPs,0.2μLEasyTaqDNAPloymerase,15.5μLddH2O。PCR反应条件为:94℃预变性5min;94℃变性40s,56℃退火30s,72℃延伸40s,34个循环;72℃延伸10min。选取阳性克隆提取质粒进行测序、鉴定[19]。
1.2.2荧光双分子互补表达载体pGN-AC4的构建参考已报道的SLACMV序列(GenBank:KT-861468.1),人工合成两端分别添加荧光双分子表达载体p1300-GN1限制性核酸内切酶MluⅠ和KpnⅠ位点两侧序列的目的片段BiFC-AC4(表1)。利用无缝克隆试剂盒将目的片段与经限制性核酸内切酶MluⅠ和KpnⅠ进行双酶切后的含绿色荧光蛋白N端159个氨基酸的植物表达载体pGN1进行连接反应,连接产物转入大肠杆菌菌株DH5α感受态细胞中,将其涂布于含卡那霉素(50mg/L)的LB平板上,于37℃,倒置培养12h。最后挑取3~5个单菌落,使用AC4特异引物AC4-1F和AC4-250R(表2)对其进行PCR鉴定,PCR反应体系(20μL)与1.2.1相同,选取阳性克隆质粒进行测序、鉴定。
1.2.3酵母自激活检测参照产品说明书进行酵母表达载体的共转化,具体如下:取100μL冰上融化的酵母AH109菌株感受态细胞,依次加入预冷的酵母猎物表达载体pGADT7-AC4、酵母诱饵表达载体pGBKT7,10μL变性后的CarrierDNA(95~100℃,5min,快速冰浴,重复1次),500μLPEG/LiAc并吸打幾次混匀,30℃水浴30min(15min时翻转6~8次混匀)。置于42℃水浴15min(7.5min时翻转6~8次混匀)。5000r/min离心40s,弃上清液,400μLddH2O重悬,离心30s,弃上清液。50μLddH2O重悬后,取15μL涂布于SD/-Trp/-Leu培养基上,置于28℃恒温培养箱中培养48~96h。挑取经PCR鉴定的单菌落进行梯度稀释,等体积同时点板于SD/-Leu/-Trp(SD-LW)和SD/-Ade/-His/-Leu/-Trp(SD-LWHA)营养缺陷型培养基中,观察并记录菌落生长以判断是否具有自激活活性。
1.2.4木薯花叶病毒AC4与PARN蛋白互作验证参照AH109ChemicallyCompetentCell产品说明书将诱饵质粒pGADT7-AC4与质粒pGBKT7-PARN共转化酵母AH109感受态细胞后,涂布于SD/-Trp/-Leu培养基上,置于28℃恒温培养箱,倒置培养48~96h,观察酵母菌落的生长情况。待长出菌落后,使用枪头挑取经PCR鉴定的单菌落于25μL无菌水中,然后再将其分别稀释10、100、1000倍,各取2μL不同浓度稀释的菌液分别点板于SD/-Trp/-Leu、SD/-Trp/-Leu/-His/-Ade培养基,置于28℃恒温培养箱,倒置培养3~5d,观察并记录木薯花叶病毒AC4与PARN是否互作[20-21]。
1.2.5荧光双分子互补验证AC4与PARN蛋白互作采用BiFC方法,注射本氏烟草进行蛋白互作检测。根据GV3101ChemicallyCompetent产品说明书将重组质粒pGN-AC4、pPARN-GC、pGN-P28和pPTN-P19分别转化到农杆菌GV3101感受态细胞内,涂布于含相同抗生素(利福平、链霉素、卡那霉素)的LB平板中,倒置放于28℃培养箱中,培养2~3d。待LB培养基长出菌落后,挑取单菌落于LB液体培养基中(含利福平、链霉素、卡那霉素),置于28℃、200r/min恒温摇床中震荡培养16~20h。配制200mL缓冲液:2mL1mmol/LMgCl2、2mL1mol/L2-吗啡乙磺酸(MES)、200μL100mmol/L乙酰丁香酮,最后用超纯水定容至200mL。将培养过夜的菌液取出以7000r/min离心15min,弃上清液,收集菌体用10mL缓冲液重悬,使用紫外分光光度计检测每种菌液OD600值,将菌液浓度稀释至OD600值为0.5。取等体积的pPARN-GC、pPTN-P19与pGN-AC4或pGN-P28的3种菌液,混匀后用无针头注射器缓慢将其分别注入3株本氏烟草中,每株注射3片叶,注射面积约为4cm2,置于培养房黑暗培养24h后,继续光照培养96h,从每株本氏烟草中各取2片叶进行激光共聚焦荧光显微镜(尼康FV1000)观察并拍照,GFP激发光为488nm[22]。
2结果与分析
2.1酵母表达载体pGADT7-AC4的构建
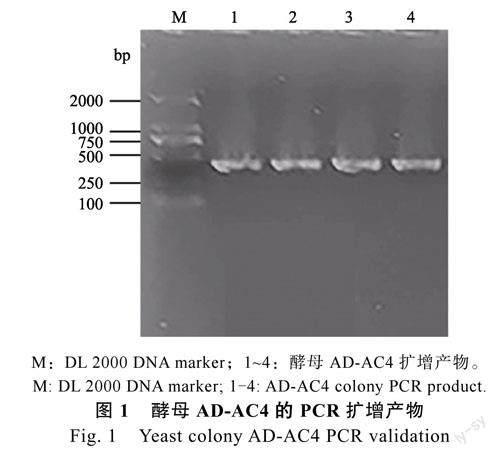
使用通用引物pGADT7-F和pGADT7-R进行鉴定,如图1所示,泳道1~4均为pGADT7-AC4(AD-AC4)不同菌落PCR产物的电泳结果,产物大小约为450bp,与预期大小一致,均为阳性克隆。选取2个阳性克隆的质粒进行测序鉴定,测序结果比对正确,说明酵母表达载体pGADT7-AC4构建成功。
2.2SLACMVAC4荧光双分子表达载体构建
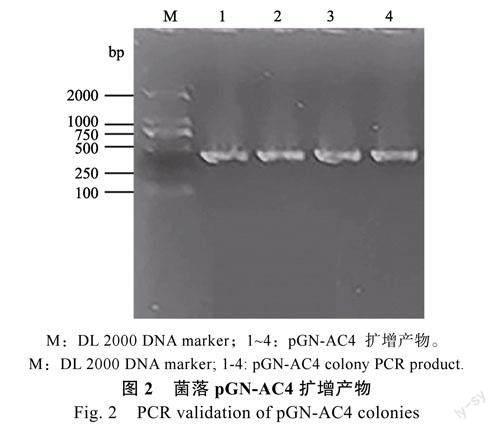
使用AC4特异引物AC4-1F和AC4-250F进行鉴定,如图2所示,泳道1~4均为pGN1-AC4(pGN-AC4)菌落PCR产物的鉴定结果,产物大小约为250bp,与预期大小一致,均为阳性克隆。选取2个阳性克隆质粒进行测序鉴定,测序结果比对均正确,说明双分子荧光互补表达载体pGN-AC4构建成功。
2.3酵母自激活验证
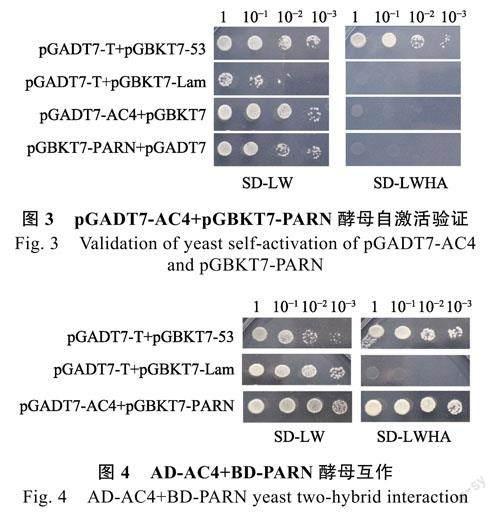
将pGADT7-AC4+pGBKT7、pGADT7+pGBKT7-PARN分别共转化酵母AH109感受态细胞,培养共转化的酵母菌落后,从SD/-Leu/-Trp(SD-LW)营养缺陷型平板中挑取单菌落,溶于25μL无菌水中,按浓度梯度稀释为10–1、10–2、10–3,梯度稀释后等体积同时点板于SD/-Leu/-Trp(SD-LW)和SD/-Ade/-His/-Leu/-Trp(SD-LWHA)营养缺陷型培养基中。以同样稀释浓度分别共转化pGADT7-T+pGBKT7-53、pGADT7-T+pGBKT7-Lam的酵母AH109,分别为阳性对照和阴性对照。结果表明pGADT7-AC4与pGBKT7、pGADT7与pGBKT7-PARN共转化的AH109酵母菌株、阳性对照和阴性对照均能在SD/-Leu/-Trp(SD-LW)营养缺陷型培养基中生长,而在SD/-Ade/-His/-Leu/-Trp(SD-LWHA)营养缺陷型培养基中只有阳性对照能生长。说明酵母表达载体pGADT7-AC4和pGBKT7-PARN均无自激活活性(图3)。
2.4木薯花叶病毒AC4与AtPARN蛋白互作
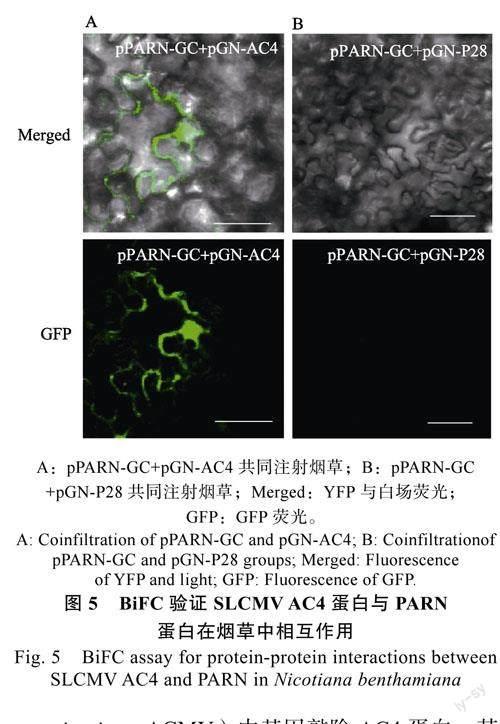
将经验证无自激活活性的AD-AC4和pGBKT7-PARN(BD-PARN)共转化的酵母菌落,从SD/-Leu/-Trp(SD-LW)营养缺陷型平板中挑取单菌落,溶于25μL无菌水中,按浓度梯度稀释为10–1、10–2、10–3,点板于SD/-Leu/-Trp(SD-LW)、SD/-Ade/-His/-Leu/-Trp(SD-LWHA)营养缺陷型培养基中,3d后观察拍照,发现AD-AC4与BD-PARN在SD/-Ade/-His/-Leu/-Trp(SD-LWHA)营养缺陷型培养基上均能生长,说明其具有相互作用(图4)。
2.5BiFC验证AC4与PARN蛋白互作
在酵母双杂交试验中验证了木薯花叶病毒AC4蛋白与PARN蛋白具有相互作用。采用BiFC方法进一步验证其互作的真实性。将菌液OD600值均为0.5的pPARN-GC、pPTN-P19与pGN-AC4或pGN-P28的农杆菌按1∶1∶1比例混匀,室温放置2h,分别共浸润3株本氏烟草,每株注射3片叶片,浸润96h后使用激光共聚焦显微镜(FV1000)观察,结果发现pGN-AC4+pPARN-GC农杆菌浸润的烟草叶片在激发光488nm均可恢复荧光,对照组pPARN-GC和pGN-P28未恢复荧光(图5),结果表明,斯里兰卡木薯花叶病毒AC4蛋白与拟南芥中的NMD关键因子PARN在本氏烟草中具有相互作用。
3讨论
双生病毒是世界上最具破坏性的植物病毒之一,对具有经济和生存重要性的作物造成严重损害[23]。由双生病毒引起的木薯花叶病,通过烟粉虱为媒介进行传播[24]。2018年在我国首次报道,SLCMV编码的AC4蛋白是一种致病因子,在SLCMV抵御寄主NMD中发挥着重要作用。有研究发现,从非洲木薯花叶病毒(Africancassavamosaicvirus,ACMV)中基因敲除AC4蛋白,其致病性被消除,表明该蛋白是ACMV病毒感染所必须的蛋白[25]。病毒是专性寄生生物,必须逃避或耐受寄主细胞的降解才能成功感染[14]。
无义介导的mRNA降解(NMD)不仅是真核生物重要的mRNA数量、质量调控机制,还是真核生物普遍存在的抗病毒防御机制[26]。因此,为初步解析SLCMV抵御NMD介导的抗病毒防御机制,本研究人工合成了AC4基因,并构建了酵母表达载体pGADT7-AC4和双分子荧光互补表达载体pGN-AC4,首先采用酵母双杂交技术证明AC4蛋白与NMD信号通路的关键因子PARN相互作用,通过BiFC技术进一步验证AC4蛋白与PARN之间的相互作用,其在烟草中均能恢复GFP的绿色荧光,通过对酵母双杂交和BiFC的试验结果,分析发现PARN与AC4蛋白具有很强的相互作用。从酵母到高等真核生物的各种生物中,去腺苷酸化是許多mRNAs降解的第一步,也是限速步骤[27]。REVERDATTO等[28]发现AtPARN对于拟南芥的胚胎发生是不可缺少的,AtPARN在多种植物脱腺苷酶中具有独特的、非冗余的作用。3ʹ核糖核酸外切酶——聚腺苷酸特异性核糖核酸酶[poly(A)-specificribonuclease,PARN]可以高效降解真核生物mRNA的聚腺苷酸尾。PARN不仅在降解mRNApoly(A)尾中发挥着关键作用,还参与了非编码RNA的加工成熟以及肿瘤等疾病过程[29-30]。有研究表明,NMD相关因子upf1、upf2和upf3X能与去帽酶DCP2、外切酶复合体(exosome)PM/Scl100和PARN等因子相互作用,并下调PARN后包含无意义密码子的mRNA(nonsensecontainingmRNA)含量增高[31]。MARAGOZIDIS等[32]发现,PARN在急性淋巴细胞白血病中的mRNA和蛋白水平明显升高,并且PARN蛋白能被磷酸化。磷酸化的PARN还能与eIF4E竞争性结合mRNA的5ʹ-帽端,从而降解mRNA[29]。丙型肝炎病毒(HepatitisCvirus,HCV)编码NMD抑制蛋白,影响EJC复合体的完整性[33]。有研究表明,花椰菜花叶病毒(Cauliflowermosaicvirus,CaMV)的病毒翻译反式激活因子TAV蛋白可作为NMD的抑制子,通过与NMD相关因子VARICOSE互作抑制NMD的降解功能[34]。推测SLCMV编码蛋白AC4不仅是RNA沉默抑制子,而且还可能通过与PARN相互作用抑制寄主NMD信号通路的抗病毒防御功能,从而提高致病性。
根据本研究发现的斯里兰卡木薯花叶病毒AC4与AtPARN蛋白的相互作用,推测SLCMV编码蛋白AC4可能通过与NMD相关因子PARN的相互作用来增强自身病毒侵染力,从而抑制其抗病毒防御功能。为证实此假说,可以使用免疫共沉淀、蛋白定位和蛋白共定位等方法对互作蛋白基因进行进一步验证,或采用过量表达NMD相关因子PARN的方法研究互作蛋白在SLCMV侵染过程中的生物学功能,进一步明确SLCMV编码蛋白AC4与寄主相互作用的分子机制。SLCMV编码的AC4蛋白与寄主蛋白如何互作?AC4如何通过与寄主蛋白互作来发挥其抑制NMD功能?这些问题均有待进一步研究。
