TLR4抑制剂预处理对脓毒性心肌病小鼠心肌损伤的改善作用及其机制研究
2024-01-27陈诗阳王晓景董玉杰张美春
陈诗阳,王晓景,董玉杰,张美春
脓毒性心肌病(sepsis-induced cardiomyopathy,SIC)是由脓毒症(sepsis)引起的发生在脓毒性休克早期的可逆性心肌功能障碍[1],其为脓毒症最严重的并发症之一。最新研究表明脓毒症伴有心功能障碍的患者病死率可高达70%,不伴有心功能障碍的脓毒症患者病死率仅为20%[2]。机体失控性炎性反应学说被认为是脓毒症发病机制的重要基础。炎性细胞过度激活并释放大量炎性细胞因子是脓毒症发生发展的重要机制之一[3],其中cTnI、CK-MB、BNP等心肌损伤标志物以及 TNF-α、IL-6、IL-1β等炎性因子在临床常用且敏感性高,是脓毒症患者疾病严重程度和预后的明确指标[4-5]。Toll样受体4 (TLR4)/NF-κB信号通路在参与脓毒性心肌病调节中起着至关重要的作用。脓毒症过程中,TLR4参与介导了NF-κB 信号途径的激活,从而大量释放TNF-α、IL-6、IL-1等炎性因子,对其后的级联反应起引领作用,进而引起心肌功能障碍[6]。本研究旨在探讨TLR4抑制剂对脂多糖(LPS)诱导SIC小鼠心肌损伤的改善作用,并进一步了解其潜在机制,报道如下。
1 材料与方法
1.1 材料 (1)实验动物:SPF级C57BL/6小鼠9只,雄性,6~8周龄,购自湖北省实验动物研究中心[生产许可证号:SCXK(鄂)2020-0018,使用许可证号:SYXK(鄂)2018-0045]。在光照 12 h / d、湿度 50%~65%、温度20~24℃的环境下喂养 1 周。(2)试剂与设备:LPS、TLR4抑制剂(TAK-242)购自美国Sigma公司;cTnI、CK-MB、BNP、TNF-α、IL-1β、IL-6 ELISA 试剂盒均购自江苏酶免实业有限公司;兔多抗TLR4、兔多抗NF-κB购自江苏亲科生物研究中心有限公司;兔多抗GAPDH购自杭州贤至生物有限公司;BCA蛋白浓度测定试剂盒购自广州捷倍斯生物科技有限公司;聚偏二氟乙烯(PVDF)膜购自美国Millipore公司;HRP标记羊抗兔二抗购自武汉博士德生物工程有限公司。 (3)仪器设备:显微镜(尼康Fi3型生物显微镜),脱水机(武汉俊杰JT-12J电脑生物组织脱水机),化学发光成像系统购自杭州申花科技有限公司,冷冻高速离心机购自湖南可成仪器设备有限公司,电热恒温培养箱购自日本ASONE,小动物超声成像系统购自百胜公司。
1.2 实验方法 本研究经武汉科技大学医学伦理委员会批准(2023102),于2023年3—6月在武汉科技大学动物实验中心进行实验。将小鼠采用随机数字表法分为对照组、SIC组、TLR4抑制剂干预组(干预组),每组3只。除对照组外,其余2组小鼠以LPS(15 mg/kg)腹腔注射构建脓毒性心肌病小鼠模型;干预组在腹腔注射LPS前1 h,经尾静脉以0.3 mg·kg-1·h-1速度静脉泵入TLR4抑制剂;对照组腹腔注射生理盐水(15 mg/kg)代替LPS。采用小动物超声成像系统测量小鼠24 h左心室射血分数(left ventricular ejection fraction,LVEF),LVEF<50%证明SIC小鼠模型构建成功[7]。
1.3 观测指标与方法
1.3.1 小鼠心肌组织形态学变化:采用HE染色观察。造模完成24 h后,颈椎脱臼法处死小鼠,取小鼠左心室心室肌组织,首先对组织进行脱水、透明、浸蜡处理,12 h后从生物组织脱水机中取出,再次进行包埋、切片烤片以及脱蜡处理,再将切片行苏木精—伊红(hematoxylin-eosin,HE)染色处理,风干后中性树胶封片,在显微镜下观察小鼠心肌组织的形态学变化。
1.3.2 血清心肌损伤和炎性标志物检测:采用酶联吸附实验(ELISA)法检测。小鼠造模24 h后,异氟烷吸入麻醉,从心脏取血1.0~1.5 ml,离心留取血清标本。使用cTnI、CK-MB、BNP、TNF-α、IL-1β、IL-6 ELISA 试剂盒,严格按照厂家说明书检测上述标志物含量。在酶标仪450 nm波长处测量每孔的光密度,并计算相应的浓度。
1.3.3 心肌组织TLR4、NF-κB蛋白表达检测:采用蛋白质免疫印迹试验(Western Blot)法检测。将少量剪碎的小鼠心室肌组织块置于冰上30 min充分裂解,4℃下离心,留取上清分装于0.5 ml离心管中并置于-20℃保存。采用BCA法测定蛋白浓度,经电泳后转移至PVDF膜上,随后封闭,封闭完成后加入兔多抗GAPDH、兔多抗TLR4、兔多抗p65(均1∶1 000稀释),4℃孵育过夜。第二日在洗去一抗液后,HRP标记羊抗兔二抗以1∶10 000稀释,室温摇床孵育2 h后再次洗去多余二抗,随后将预混好的免疫荧光(ECL)发光液滴加于PVDF膜上,反应数分钟待荧光带明显后,用滤纸吸去多余的底物液,覆上保鲜膜,X线胶片压片后依次放入显影液显影、定影液定影,冲洗、晾干、扫描胶片,用ipp6.0软件分析胶片灰度值。

2 结 果
2.1 各组小鼠心肌组织病理形态变化 在各组小鼠造模完成24 h后显微镜下可见: 对照组心肌组织形态结构完整,心肌纤维排列规则,心肌细胞整齐未见明显病变,未见炎性细胞浸润; SIC组心肌组织形态结构相对完整,心肌纤维排列紊乱,部分心肌细胞水肿,胞体崩解消融(黑色箭头),间质内可见明显炎性细胞浸润(黄色箭头); 干预组心肌组织形态结构相对完整,心肌纤维排列规则,少量心肌细胞损伤伴有空泡样变(黑色箭头),间质内可见零散炎性细胞浸润(黄色箭头),见图1。
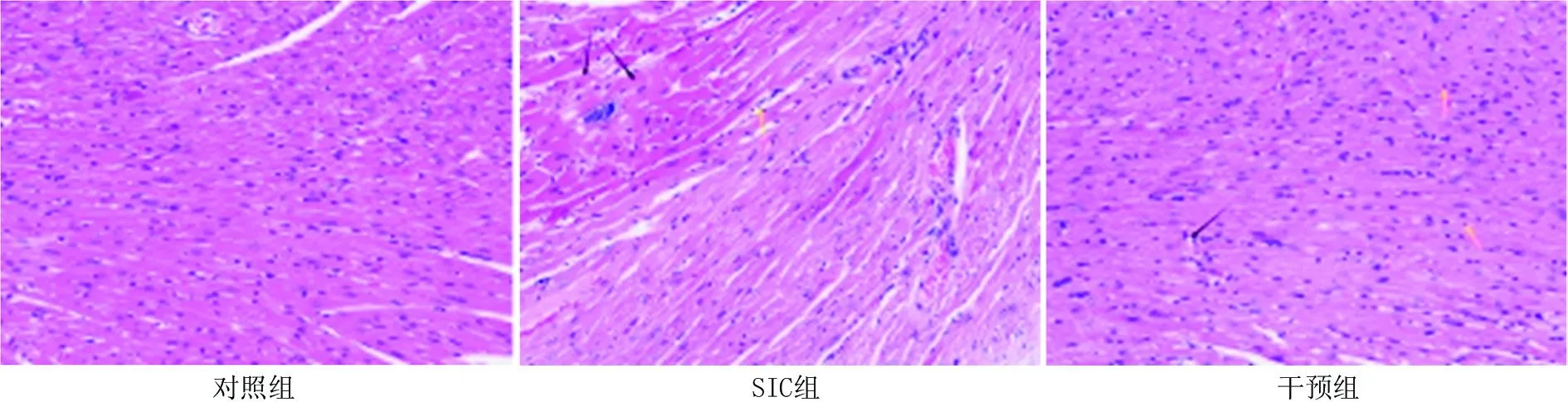
图1 各组大鼠心肌组织病理形态改变( HE 染色,×200)Fig.1 Pathological changes in myocardial tissue of rats in each group under light microscopy (HE staining, × 200)
2.2 各组血清心肌损伤标志物及炎性因子水平比较 在小鼠造模24 h后,与对照组比较,SIC组cTnI、CK-MB、BNP、TNF-α、IL-1β、IL-6表达水平均升高(t=26.274、32.979、26.782、36.031、23.266、34.881,P均<0.001);与SIC组比较,干预组cTnI、CK-MB、BNP、TNF-α、IL-1β、IL-6表达水平均下降(t=15.181、22.537、20.697、21.428、19.241、22.228,P均<0.001),见表1。
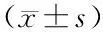
表1 3组小鼠血清心肌组织损伤标志物及炎性因子水平比较Tab.1 Comparison of serum myocardial tissue injury markers and inflammatory factor levels among three groups of mice
2.3 各组心肌组织TLR4、NF-κB蛋白表达比较 在小鼠造模24 h后,与对照组比较,SIC组TLR4、NF-κB蛋白表达显著上调(t=15.611、9.977,P均<0.001),与SIC组比较,干预组TLR4、NF-κB表达下调(t=28.067、7.320,P均<0.01),见图2、表2。
表2 3组小鼠心肌组织TLR4、NF-κB蛋白表达比较Tab.2 TLR4 and NF in myocardial tissue of three groups of mice-κB Comparison of protein expression

图2 各组小鼠心肌组织TLR4、NF-κB蛋白印记图Fig.2 TLR4 and NF in myocardial tissue of mice in each group-κB protein blot
2.4 血清炎性因子水平与心肌组织TLR4、NF-κB蛋白表达的相关性 进一步行相关性研究分析示,炎性因子TNF-α、IL-1β、IL-6的表达水平与小鼠心肌组织中TLR4、NF-κB蛋白表达呈高度正相关(P<0.01),见表3。
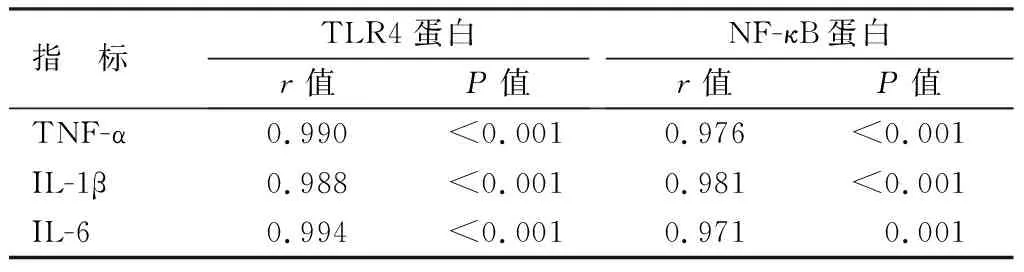
表3 炎性因子水平与TLR4、NF-κB蛋白表达的相关性Tab.3 Inflammatory factor levels and TLR4, NF-κB The correlation between protein expression
3 讨 论
关于脓毒性心肌病的有效治疗手段,至今仍是国内外悬而未决的一大难题。近年来,关于脓毒性心肌病的发生发展机制已有了一定的深入研究,其中由于炎性因子的过度表达而引起一系列的恶性循环基本取得了专家们的共识。因此在关于脓毒性心肌病的治疗上,除了重视引起脓毒症感染的原发病以外,抗炎治疗现已成为首要的治疗原则[8]。由于在发生脓毒性心肌病时,机体会释放大量炎性因子,如果仅仅只是通过针对单一炎性因子的抑制来达到抗炎的目的,显然是不足以改善机体发生的炎性瀑布反应。治疗脓毒性心肌病的干预靶点应着眼于调节炎性因子的关键部位。目前脓毒性心肌病药物治疗效果不理想,研究者认为这可能与现阶段治疗脓毒性心肌病药物的靶点共性不高,对于早期炎性因子表达的抑制作用较差相关。因此,在现有的介导炎性因子表达的信号通路上,探索各信号传导通路之间的交互作用,在不同信号通路的交叉点上开发靶点药物,为治疗脓毒性心肌病提供新思路。
TLR4/NF-κB信号通路的异常调控与脓毒症、缺血再灌注损伤、癌症、消化性溃疡、炎性肠病、肥厚性心肌病、COVID-19等多种疾病相关[9-15]。炎性因子的大量释放也与TLR4信号通路的表达、激活密不可分。TLR4/NF-κB信号通路是脓毒性心肌病发生、发展、转归的一个重要的信号通路[16],当机体收到LPS刺激后,LPS与脂多糖结合蛋白(LBP)结合并转移到细胞表面的CD14上。然后,TLR4作为一种Ⅰ型跨膜蛋白与LPS结合后再与辅助蛋白MD-2相互作用,激活下游各种信号传导通路,包括NF-κB、JNK、p38MAPK以及PI3K/AKT信号通路[17],进而导致炎性因子的大量释放引起心肌损伤。因此TLR4作为各传导通路的共同上游信号通路,标志着TLR4可能成为有效治疗脓毒性心肌病的新靶点。TLR4抑制剂作为治疗某些疾病的药物,纠正TLR4信号通路过度激活的主要方法有3种:包括下调TLR4的表达;直接与髓样分化复合物2(MD2)结合抑制TLR4信号通路;直接与TLR4或TLR4/MD2复合物结合抑制TLR4活性[18]。在研究TLR4抑制剂干预脓毒症的相关作用机制方面,国内外学者均做了大量的工作。Selfridge等[19]设计了一些纳曲酮衍生物,构效关系表明,用纳曲酮可以显著提高TLR4的抑制活性。刘新强等[20]通过实验得出TLR4抑制剂E5564可通过与TLR4结合并抑制其与LPS的结合,阻断LPS-TLR4信号通路的激活,进而减轻心脏炎性反应,改善心功能。杨梦等[21]提出TLR4特异性抑制剂TAK-242是通过抑制TLR4/NF-κB通路的激活,从而减轻脓毒症大鼠肝脏的炎性反应。Achek等[22]在小鼠实验中证实磷脂酰肌醇二磷酸(PIP2)通过特异性结合TLR4上的MD2结合位点,从而抑制TLR4/MD2复合物的形成,进一步抑制LPS诱导的多种促炎因子的产生。
LPS常用于建立以心室扩张、左心室射血分数降低和心功能不全为特征的脓毒症模型。本研究发现,LPS处理组(SIC组+干预组)小鼠血清cTnI、CK-MB、BNP、TNF-α、IL-1β、IL-6水平均显著升高,且与TLR4、NF-κB蛋白表达呈正相关。经TLR4特异性抑制剂TAK-242干预后,上述损伤标志物及炎性因子较SIC组均明显下降,进一步证实了该型抑制剂可通过下调TLR4、NF-κB蛋白表达有效降低心肌损伤标志物以及炎性因子的释放,进而改善心功能。
综上所述,TLR4抑制剂干预可改善LPS所致的心肌损伤,其机制推测与其下调TLR4/NF-κB信号通道蛋白表达从而抑制炎性反应相关。因此TLR4作为治疗脓毒性心肌病一个重要的靶点,设计和开发针对这一靶点的抑制性药物具有很高的治疗潜力。
利益冲突:所有作者声明无利益冲突
作者贡献声明
陈诗阳:设计研究方案,实施研究过程,撰写论文;王晓景:提出研究思路,设计研究方案;董玉杰:协作实施研究过程;张美春:提出研究方向、研究选题,论文审核
