基于生物信息学构建口腔鳞状细胞癌免疫基因的预后模型
2024-01-27王锦航彭士雄杨凯成陈彦平崔子峰
王锦航,彭士雄,杨凯成,陈彦平,崔子峰
口腔鳞状细胞癌(oral squamous cell carcinoma,OSCC)是头颈部最常见的恶性肿瘤。尽管临床诊断和治疗不断改进,但过去10年间OSCC患者的病死率并未显著改善[1-2]。目前临床中用于风险分层或预后监测的传统指标在评估OSCC患者的预后中仍存在许多不足,因此有必要挖掘潜在的生物标志物来更准确地实现对OSCC患者预后的评估,从而帮助临床医生为不同风险的OSCC患者选择个性化的治疗策略。
免疫细胞广泛分布于肿瘤免疫微环境(tumor immune microenvironment,TIME)中,并与肿瘤的发生和进展密切相关[3]。免疫逃逸可通过表达免疫监视位点配体或分泌免疫抑制因子来抑制免疫系统对肿瘤的有效识别和杀伤,被认为是肿瘤的特征之一[4-5]。既往学者的研究已证实了单个或多个免疫相关基因(immune-related genes,IRGs)在OSCC中的表达失调,但缺乏结合数据库分析IRGs与TIME或OSCC患者预后间关系的研究。因此,本研究利用癌症基因组图谱(the cancer genome atlas,TCGA)-OSCC数据库和免疫基因数据库等,构建了一个由15个预后相关且差异表达的免疫相关基因(differentially expressed immune-related genes,DEIRGs)组成的OSCC风险预测模型,并探讨了该模型在OSCC患者预后和临床中的价值。此外,本研究还分析了DEIRGs的转录调控机制以及模型对临床病理和TIME的影响,报道如下。
1 资料与方法
1.1 数据的获取和处理 本研究使用R软件(v3.5.2)进行统计分析和绘图。从TCGA数据库下载截至2021年7月HTSeq-FPKM类型的OSCC RNA-seq数据,包括330个OSCC和32个正常样本。数据中的基因名称通过Ensembl数据库提供的文件“homo_sapiens.grch38.104.chr.gtf.gz”进行注释和转换。最后通过R“limma”包对数据进行预处理。OSCC患者的预后和临床病理数据同样从TCGA数据库下载,其中包括性别、年龄、总生存期(overall survival,OS)、生存状态、肿瘤分化程度、肿瘤分期和T分期。由于数据中大多数OSCC患者的M和N分期数据缺失,因此本研究未包括这些数据的分析。IRGs目录从ImmPort数据库下载,包含2 483个已通过研究验证的IRGs。与癌症相关的318个转录因子(transcription factors,TFs)目录从Cistrome癌症项目下载。
1.2 DEIRGs的鉴定和富集分析 对预处理后的数据进行Wilcoxon秩和检验,以FDR<0.05和|logFC|>1作为阈值来鉴定OSCC和正常样本间的差异表达基因。将差异表达基因与IRGs间的重叠基因选定为DEIRGs。通过R包(clusterProfiler、org.hs.eg.db、enrichplot、ggplot2)、Gene Ontology(GO)和Kyoto Encyclopedia of Genes and Genomes(KEGG)数据库对DEIRGs行富集分析,以明确DEIRGs可能参与的生物学机制,P<0.05为差异具有统计学意义。
1.4 预后相关DEIRGs和差异表达转录因子(diffe-rentially expressed TFs,DETFs)间调控网络的构建 选择在TFs和差异表达基因间重叠的基因作为DETFs。以P<0.05和|COR|>0.3作为阈值,利用Pearson检验分析预后相关DEIRGs和DETFs间的关系。Cytoscape软件(v3.8.2)可视化两者间的调控网络。
1.5 风险预测模型的能力和相关性分析 依据模型公式计算每个OSCC患者的RiskScore,并以中位数作为截断值将其分为低风险组和高风险组。使用R“survivor”包对低、高风险组行Kaplan-Meier生存曲线分析。使用R“survivalROC”包绘制时间相关的受试者工作特征(ROC)曲线,并计算曲线下面积(AUC)来评估模型预测预后的准确性。AUC值的范围:0.5~0.7表示中等准确性,>0.7~0.9表示较高准确性,>0.9表示高准确性[7]。使用R“survivalROC”包评估模型相较于传统临床病理特征的预测准确性优势。使用R“survivor”包对模型和OSCC患者的临床病理特征进行单因素和多因素Cox分析,以验证模型的独立预后价值。使用R“beeswarm”包对模型和OSCC患者的临床病理特征进行相关性分析。利用TIMER数据库和Pearson检验分析模型分别与B细胞、CD4+T细胞、CD8+T细胞、巨噬细胞、中性粒细胞和树突状细胞间浸润的关系。P<0.05为差异具有统计学意义。
2 结 果
2.1 差异表达基因、DEIRGs和DETFs的鉴定 在OSCC和正常样本的39 740个mRNA中共鉴定出3 634个差异表达基因(FDR<0.05和|logFC|>1),其中上调基因2 404个,下调基因1 230个(图1A)。差异表达基因和IRGs的交集鉴定出330个DEIRGs,其中上调基因215个,下调基因115个(图1B)。差异表达基因与TFs的交集鉴定出61个DETFs,其中上调基因42个,下调基因19个(图1C)。
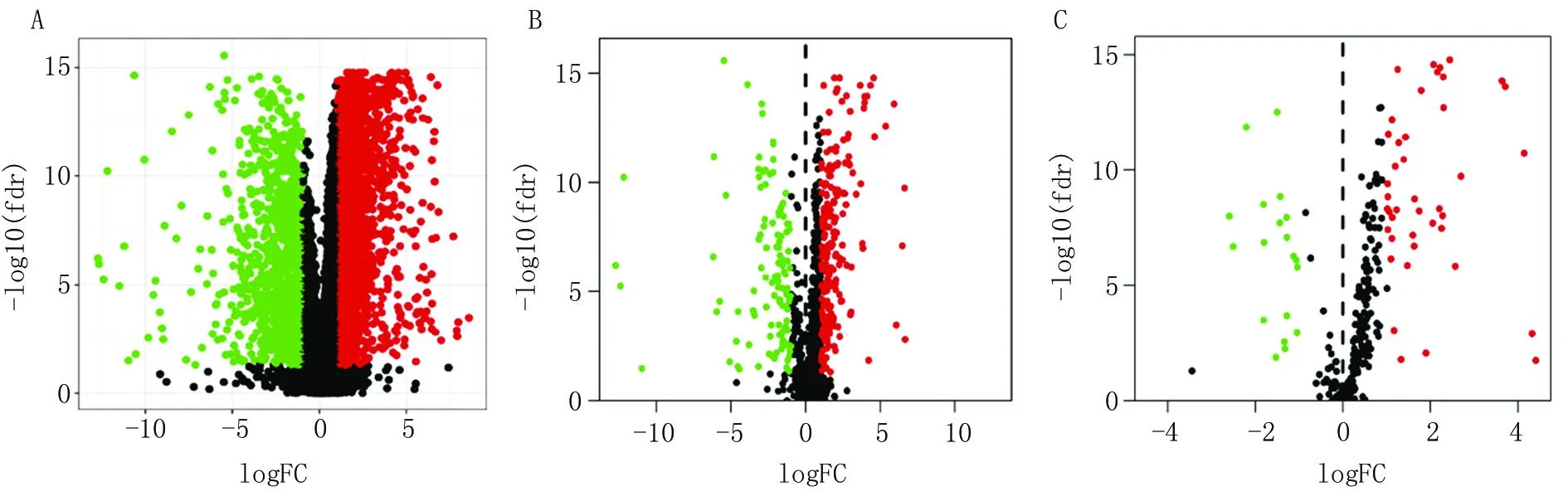
注:A. 差异表达基因的鉴定;B. DEIRGs的鉴定;C. DETFs的鉴定。红色为上调基因, 绿色为下调基因, 黑色为无差异基因。图1 差异表达基因、DEIRGs和DETFs的鉴定Fig.1 Identification of differentially expressed genes, DEIRGs, and DETFs
2.2 DEIRGs的富集分析 GO富集分析显示,DEIRGs主要涉及“GO:0048018 受体配体活性” “GO:0005126 细胞因子受体结合”和“GO:0005125 细胞因子活性”等(P<0.05)。KEGG富集分析显示,DEIRGs主要参与“hsa04060 细胞因子—细胞因子受体相互作用”“hsa04151 PI3K-Akt信号通路”和“hsa04630 JAK-STAT信号通路”等(P<0.05),见表1。
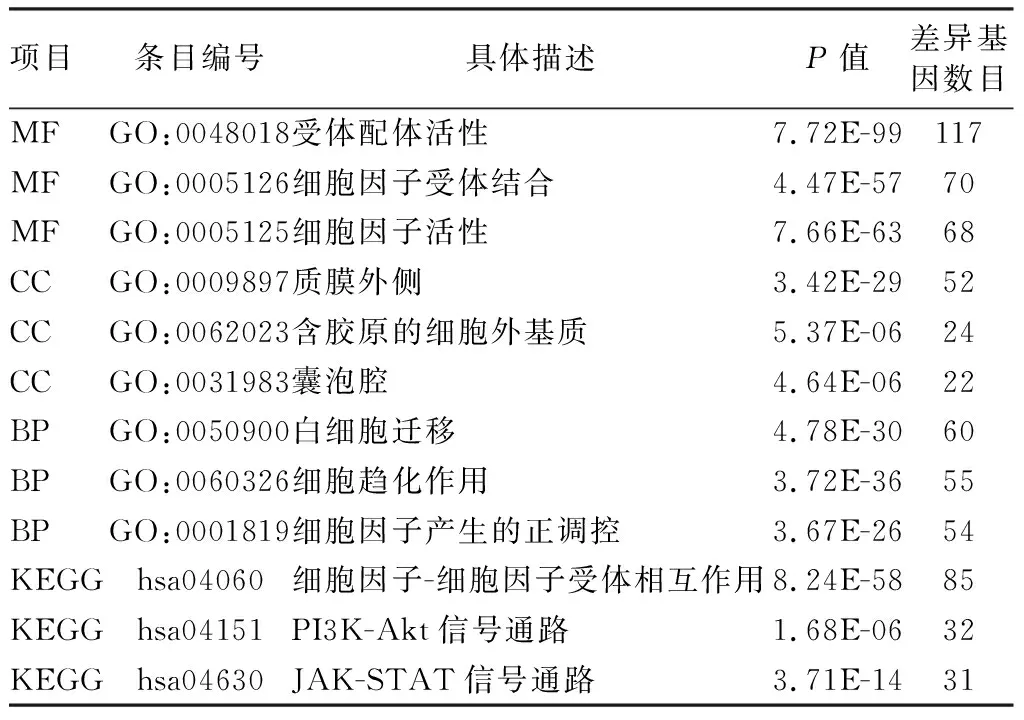
表1 DEIRGs的富集分析Tab.1 Enrichment analysis of DEIRGs
2.3 预后相关DEIRGs和DETFs间的调控网络 通过对DEIRGs和OSCC患者的OS行单因素Cox回归分析筛选出20个与预后相关的DEIRGs(P<0.05)(图2A),分别为VEGFD、SFTPA2、SEMA3A、GAST、BIRC5、STC1、DKK1、CCL26、IL1A、PLAU、CXCL8、SLURP1、PDGFRB、PGLYRP4、IL36A、TNFRSF19、CTLA4、SEMA3G、IL17RD、AVPR2。Pearson检验发现16个预后相关的DEIRGs 和28个DETFs间存在相关性(P<0.05,|COR|>0.3),其中仅SLURP1与BRCA1或CDK2之间以及BIRC5与KAT2B之间呈负相关,其余均为正相关。并且BIRC5、SEMA3G、IL17RD和CTLA4在该调控网络中可能扮演核心角色(图2B)。
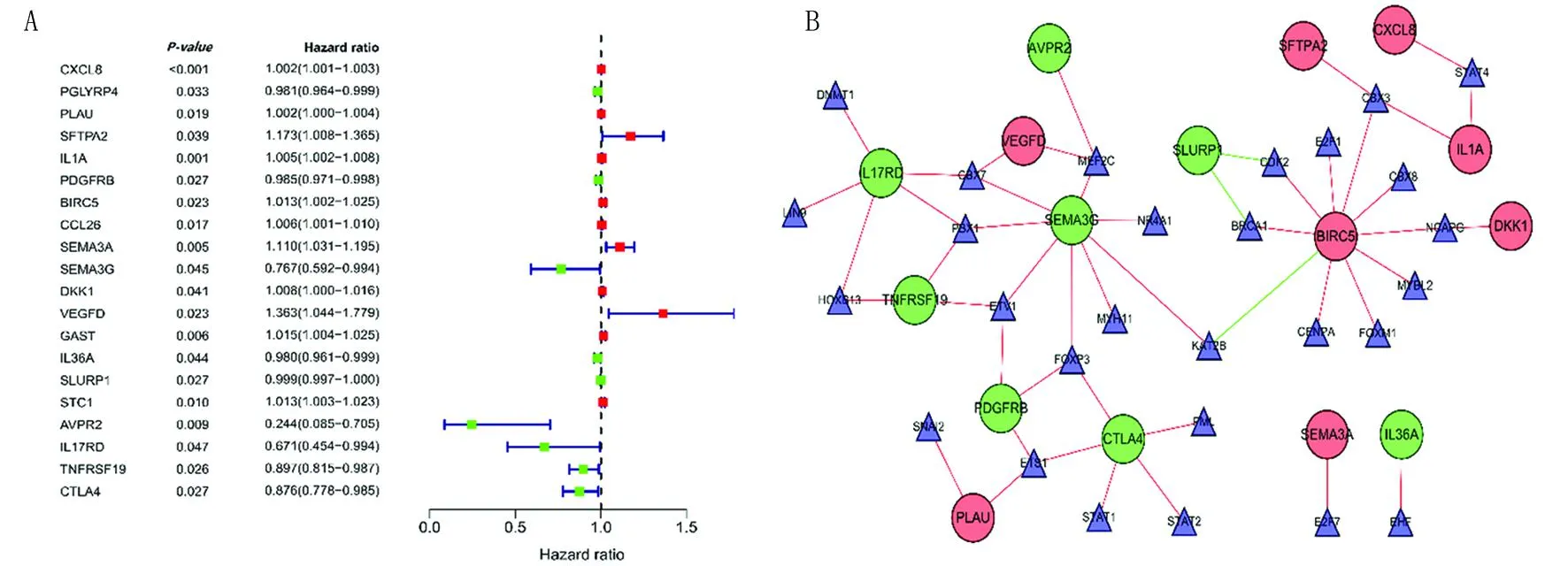
注:A. 预后相关DEIRGs的森林图。红色为高风险;绿色为低风险。B. 预后相关DEIRGs和DETFs间的调控网络。红圈为高危DEIRGs, 绿圈为低危DEIRGs ,蓝三角为DETFs;红线为正相关, 绿线为负相关。图2 预后相关DEIRGs和DETFs间的调控网络Fig.2 Regulatory network between prognosis related DEIRGs and DETFs
2.4 风险预测模型的构建 通过对20个预后相关DEIRGs行R“step”函数和多因素Cox回归分析,筛选出其中15个用于构建风险预测模型。在该模型中,VEGFD、SFTPA2、SEMA3A、GAST、STC1、CCL26、CXCL8为高风险基因(HR>1),SLURP1、IL36A、PDGFRB、BIRC5、PGLYRP4、TNFRSF19、IL17RD、AVPR2为低风险基因(HR<1)。模型计算公式为RiskScore =(0.530859772×VEGFD)+(0.208685511×SFTPA2)+(0.087666745×SEMA3A)+(0.012001686×GAST)+(0.009443492×STC1)+(0.007784836×CCL26)+(0.001318513×CXCL8)+(-0.001600693×SLURP1)+(-0.014374813×IL36A)+(-0.016585009×PDGFRB)+(-0.020523714×BIRC5)+(-0.024620582×PGLYRP4)+(-0.1003187×TNFRSF19)+(-0.475553967×IL17RD)+(-1.032424×AVPR2),见表2。
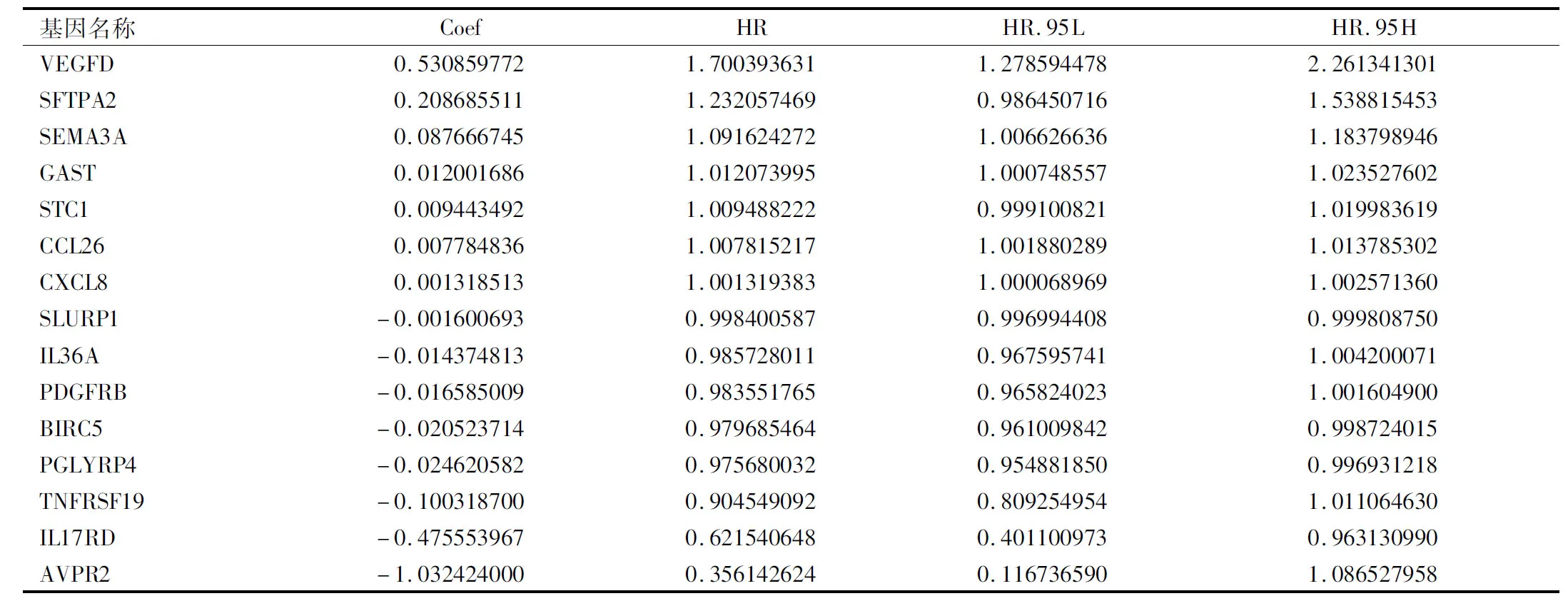
表2 OSCC风险预测模型中15个预后相关DEIRGs的多因素Cox回归分析Tab.2 Multivariate Cox regression analysis of 15 prognostic related DEIRGs in the OSCC risk prediction model
OSCC患者根据RiskScore的中位数分为高风险组(n=154)和低风险组(n=155)。Kaplan-Meier分析得出,高风险组和低风险组的5年生存率分别为0.271(95%CI0.173~0.426)和0.649(95%CI0.539~0.783),高风险组的预后明显差于低风险组(P<0.001)(图3A)。ROC曲线发现该模型的AUC值为0.732,表明其预测能力具有较高的准确性(图3B)。
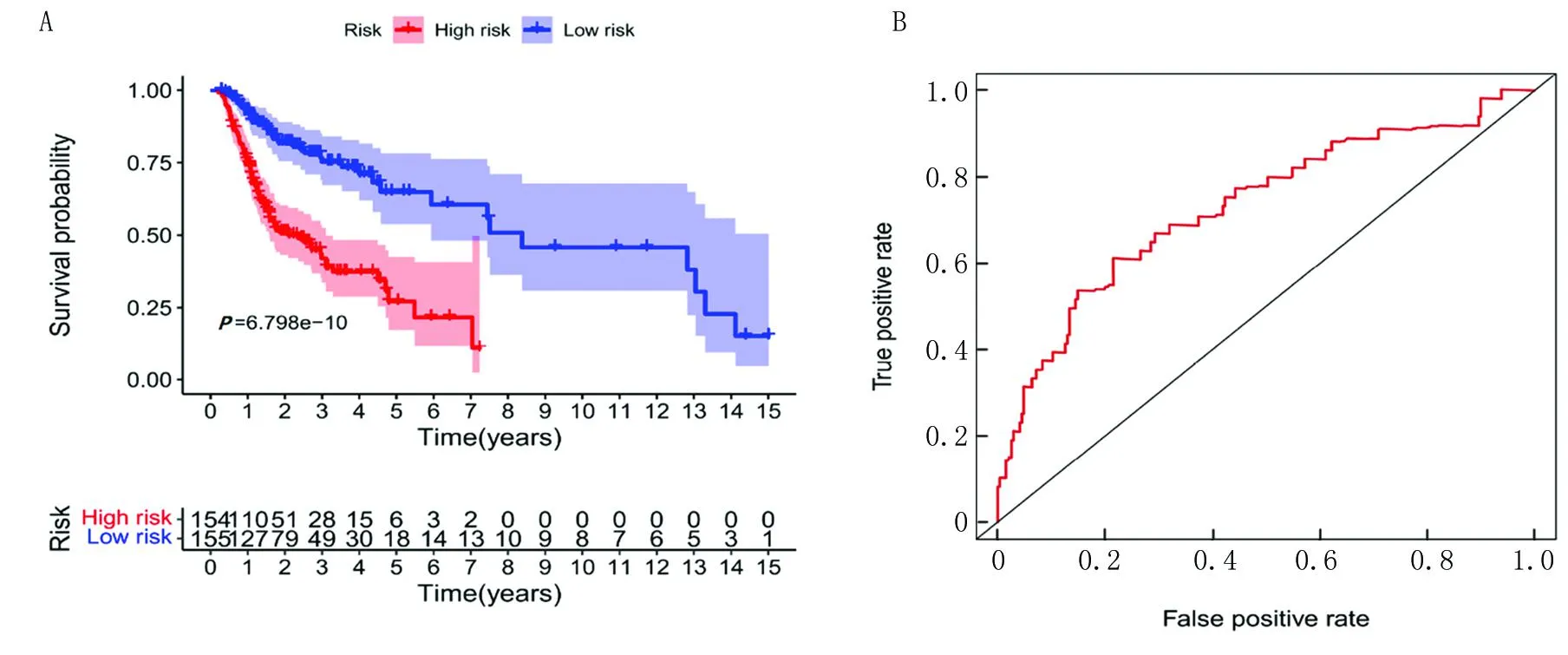
注:A. Kaplan-Meier生存分析;B. ROC曲线分析。图3 OSCC患者的风险预测模型Fig.3 Risk prediction model for OSCC patients
2.5 风险预测模型的能力分析 结合临床病理特征后行单因素Cox回归分析,结果显示年龄(P=0.008)、肿瘤分期(P<0.001)、T分期(P=0.002)和RiskScore(P<0.001)与预后相关(图4A)。进一步行多因素Cox回归分析,结果表明年龄(P=0.001)和RiskScore(P<0.001)与预后独立相关(图4B)。ROC曲线分析显示,相比于传统的临床病理特征,该模型具有更好的准确性优势(AUC=0.717)(图4C)。
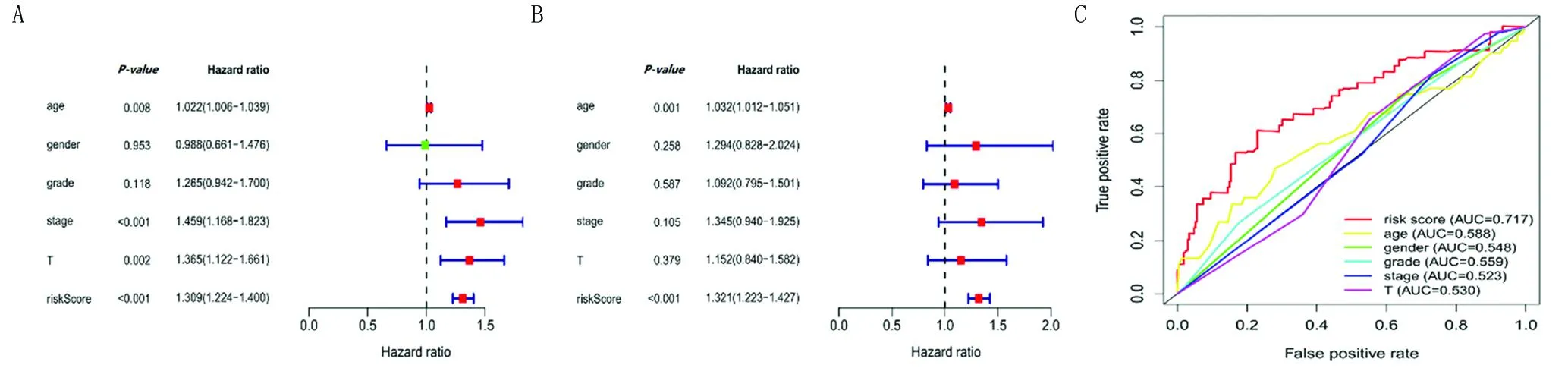
注:A:单因素Cox回归分析;B:多因素Cox回归分析。红色为高危因素;绿色为低危因素。C:ROC曲线评估模型的准确性优势。图4 风险预测模型的能力分析Fig.4 Analysis of the Capability of Risk Prediction Models
2.6 风险预测模型的相关性分析 相关性分析结果表明,男性患者中PDGFRB和BIRC5的表达水平较高;G1和G2时PGLYRP4和IL36A的表达水平较高,G3和G4时AVPR2和IL17RD的表达水平较高;Ⅲ期和Ⅳ期时CXCL8、BIRC5、SEMA3A、GAST、STC1和RiskScore的水平较高;T3和T4期时STC1的表达水平较高,AVPR2的表达水平较低(P<0.05)(图5)。这些结果表明,DEIRGs基因的异常调节与OSCC的发展密切相关。

注:A~B.性别;C~F.分化程度;G~L.肿瘤分期;M~N. T分期。图5 风险预测模型与临床病理特征的相关性Fig.5 Correlation between risk prediction model and clinical pathological features
为了确定模型是否与OSCC患者的TIME相关,利用TIMER数据库分析了RiskScore与TIME中免疫细胞浸润间的关系。Pearson检验结果发现,RiskScore 与B细胞(Cor=-0.180,P=0.002)和CD4+T细胞(Cor=-0.127,P=0.026)呈负相关,与CD8+T细胞、树突状细胞、巨噬细胞和中性粒细胞无相关性(P>0.05)(图6)。
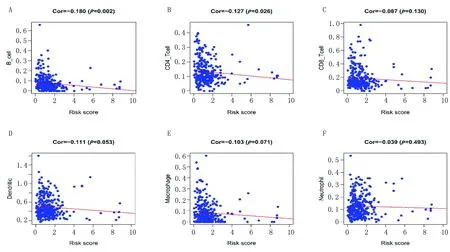
注:A. B细胞;B. CD4+T细胞;C. CD8+T细胞;D. 树突状细胞;E. 巨噬细胞;F. 中性粒细胞。图6 风险预测模型与肿瘤免疫细胞浸润的相关性Fig.6 Correlation between risk prediction model and tumor immune cell infiltration
3 讨 论
风险分层对于预测恶性肿瘤患者的预后和治疗决策至关重要。目前,对于OSCC患者预后的预测和治疗的选择主要基于美国癌症分期联合委员会(AJCC)发布的TNM分期以及美国国立综合癌症网络(NCCN)发布的肿瘤临床实践指南,但临床工作中发现即使在具有相同或相似TNM分期和治疗方式的患者中,预后也存在显著差异[8-9]。肿瘤免疫靶点治疗药物的出现改善了部分晚期OSCC患者的生存率,并且通过增强免疫应答、刺激肿瘤特异性免疫、打破免疫耐受或重新激活免疫细胞等方式识别并杀死肿瘤细胞[10-11],这种治疗方式涉及代谢、炎性反应和TIME等多种复杂机制的改变。考虑到肿瘤细胞异型性甚至癌变往往发生于炎性细胞密集浸润的TIME中[12],因此本研究期望通过探讨OSCC中免疫相关基因组和TIME的变化,提供关于预测患者预后和发现免疫治疗潜在靶点的重要依据。
本研究在OSCC的差异表达基因中共发现了330个DEIRGs。富集分析表明,其中涉及的主要生物学特征为细胞因子、JAK-STAT信号通路以及PI3K-Akt信号通路。细胞因子可以引起人体TIME或免疫系统的变化,并且在OSCC中激活的免疫细胞已被证实可通过分泌炎性细胞因子促进肿瘤细胞的免疫逃逸[13-14]。JAK-STAT信号通路参与调节免疫系统,影响肿瘤细胞的分化、增殖、凋亡[15-16]。PI3K/Akt信号通路中关键因子的异常活化可促进OSCC细胞的侵袭和转移,并与患者的肿瘤分化程度、肿瘤分期和淋巴结转移密切相关[17]。此外,本研究在DETFs和DEIRGs的调控网络中发现了28个DETFs和16个预后相关的DEIRGs之间存在调控关系,其中BIRC5、SEMA3G、IL17RD和CTLA4可能是该网络中的核心基因。因此,DEIRGs及其所涉及的生物学特征在OSCC和免疫调节中发挥着重要功能,对于分析肿瘤的发展或制定治疗措施具有参考价值。
尽管近期的研究已经确定了一些可以改善OSCC诊断或治疗的生物标志物,但由于OSCC的高复发率和不良预后,研究仍需要探索潜在的生物标志物以便精准地明确OSCC患者的预后情况,这将有助于根据每位OSCC患者的不同预后风险采取个性化的治疗措施。因此,本研究构建了一个包含15个预后相关DEIRGs的风险预测模型,该模型中的部分DEIRGs如SEMA3A、CXCL8和BIRC5等基因已经被研究证明与OSCC密切相关[18-20],这些DEIRGs具有成为免疫治疗靶点或预后生物标志物的潜力。本研究同时评估了该模型的准确性和临床价值。与低危组相比,模型中高危组患者的预后明显较差,且高危组的肿瘤分期更晚,表明该模型不仅能预测患者的预后情况,还能预测 OSCC 的进展。ROC曲线显示模型的AUC为0.732,证明该模型具有较高的诊断准确性。Cox回归分析证实该模型为OSCC患者的独立预后因素,ROC曲线进一步明确了该模型与传统的临床病理特征相比具有更好的准确性优势。
本研究还分析了模型与免疫细胞浸润的相关性,以反映OSCC中TIME的状态。研究发现RiskScore与B细胞和CD4+T细胞呈负相关,可以看出随着RiskScore的增加机体的免疫功能逐渐受损,这可能导致TIME的失衡。以上结论证实了该模型具有较高的风险分层能力,对治疗决策具有较高的指导价值。但本研究仍存在一些局限性。首先,该研究使用了来自多个数据库的大样本量数据集,但仍需进行更广泛的验证。其次,还需要进行体内和体外实验研究,以阐明模型中15个预后相关DEIRGs的具体分子调控机制。
综上所述,本研究基于DEIRGs构建了一个较为精确的风险预测模型,其中15个预后相关的DEIRGs充当重要角色。这些发现为理解OSCC的免疫机制提供了新的视角,也为临床医生选择个性化治疗策略提供帮助,以提高OSCC患者的生存率。
利益冲突:所有作者声明无利益冲突
作者贡献声明
王锦航:数据获取,统计分析,论文撰写;彭士雄:数据获取,统计分析;杨凯成:数据获取,参与撰写;陈彦平:研究构思,论文审核;崔子峰:课题设计,论文撰写,论文终审
