IgA肾病的遗传学研究进展
2024-01-24周绪杰
曾 赏,周绪杰
(北京大学第一医院肾内科,北京大学肾脏疾病研究所,卫生部肾脏疾病重点实验室,教育部慢性肾脏病防治重点实验室,北京 100034)
IgA肾病(IgA nephropathy,IgAN)是全球最常见的原发性肾小球肾炎,在中国占原发性肾小球疾病的比例高达40%~50%[1],也是慢性肾脏病和终末期肾衰竭的重要原因,20%~40%的IgAN患者在20年内进展为终末期肾病[2]。它的诊断依赖于肾活检,表现为肾小球系膜区含有IgA的免疫复合物沉积和组织病理学病变,如系膜细胞增生和细胞外基质积聚。近年来,IgAN的发病机制的主流观点多为“四重打击学说”,首先是在遗传易感性的基础上,个体在细菌或病毒感染后激发黏膜免疫反应,身体免疫应答出现失调后产生半乳糖缺陷型IgA1(Gd-IgA1),其触发异常抗聚糖抗体应答产生抗Gd-IgA1的抗糖抗体,随后形成免疫复合物并沉积在肾小球系膜中,激活补体系统和其他介质,刺激系膜细胞增殖、分泌系膜基质、细胞因子等而致肾小球炎症反应,最终导致肾小球硬化和间质纤维化,见图1。
IgAN的发病存在种族和地域的差异,非洲人群的发病率最低,欧洲人群的发病率中等,亚洲人群的发病率最高,有明显的从西到东的上升趋势[3]。其次,陆续有众多家族性IgA肾病家系的报道[4]。在大多数已报道的家系中,IgAN遵循不完全外显的常染色体显性遗传模式。越来越多的证据表明遗传因素参与了IgAN的发病和进展。既往主要运用连锁分析与候选基因关联分析的方法来分析IgN的易感基因,而在过去的十年中,全基因组关联研究(genome-wide association study,GWAS)进一步发现了与IgAN相关的遗传变异。在多个GWAS中显示出显著关联的位点主要参与抗原加工提呈、补体系统、粘膜先天免疫、粘膜IgA产生的调节、免疫应答和炎症反应等,进一步支持“四重打击学说”。这些GWAS发现的易感位点在某种程度上指明了IgAN的致病途径,肯定了人体免疫、补体激活和Gd-IgA1的产生等在IgAN发病机制中的作用。然而GWAS定位到的遗传变异多位于非编码区,并不一定是真正的致病变异,所以需要对GWAS结果进行深入挖掘,以筛选功能位点并阐明其潜在的发病机制,从而指导未来的疾病诊疗、风险预测、药物开发等在内的精准医学。此外,随着样本量的增加以及基因分型和测序成本的降低,越来越多地使用全外显子测序和全基因组测序来识别罕见变异,这可能解释复杂性状中遗传力缺失的部分。文章总结了近年来IgAN遗传学研究的现状,并对该领域的关键挑战进行了展望。
1 IgAN易感性的连锁分析和关联分析
连锁分析是在家系中检验IgAN和等位基因间的遗传模式,对中效或微效基因的定位效能不足。目前运用连锁分析发现的IgAN易感区域包括2q36、3p23-24、4q26-31、6q22-23和17q12-22,但未在这些区域中寻找到致病基因[5~7]。
关联分析是在人群中检验IgAN和等位基因间的相关性,包括有遗传假设的候选基因关联分析和无遗传假设的GWAS。候选基因关联分析是在已知病理生理基础上,选择一些可能与IgAN有关的基因分析其是否与疾病相关联,其中既往研究主要包括了免疫识别相关基因[8~9]、肾素血管紧张素系统相关基因[10]以及细胞因子相关基因[11]等。然而因样本量小,统计分析方法滞后和未在独立人群中进行验证等缺点。而GWAS是无先验假说的在人类全基因组范围内寻找与疾病关联的遗传变异。
2 IgAN易感性的GWAS研究
在过去的十多年中,GWAS逐渐揭示了IgA肾病的遗传结构。见表1[12~22]。2010年发表的在欧洲人群进行的第一次GWAS在主要组织相容性区域(HLA-B、DRB1、DQA和DQB位点)检测到一个强的全基因组显著信号,该位点对抗原的加工和呈递和适应性免疫至关重要[12]。由于受限于样本量而没有观察到MHC区域以外的关联。2011年第二个GWAS在东亚和欧洲人都证实了MHC中的关联信号,还鉴定了两个非MHC位点,包括补体因子H和HORMAD2区段[13]。CFHR1和CFHR3基因编码因子H相关肽,调节补体旁路途径的活性。研究显示FHR1与H因子竞争结合表面固定的C3b,导致C3转化酶的活性增加。CFHR1和CFHR3的保护性缺失与更高的循环中H因子水平和抑制补体旁路的局部激活有关[23]。起初定位的HORMAD2编码粘膜免疫和炎症的相关细胞因子。随后发表的在中国队列中进行的GWAS中还发现了另外两个新的重要基因座,编码与黏膜免疫相关的基因:TNFSF13和DEFA,它们分别编码促进IgA产生的强大黏膜细胞因子APRIL(B细胞活化因子)和在粘膜表面分泌的对微生物病原体提供先天防御的α-防御素人抗菌肽[14]。随后2014年发表的扩大样本的多种族GWAS分析了欧洲和东亚血统的7658例病例和12954例对照,除了验证MHC、HORMAD2、CFH、TNFSF13和DEFA位点的关联外,该研究还发现了几个新的基因座位,包括VAV3、ITGAM-ITGAX和CARD9,以及HLA和DEFA位点的新独立信号[15]。VAV蛋白是B细胞适应性免疫功能和NF-κB激活所必需的鸟嘌呤核苷酸交换因子,这一过程刺激IgA的产生[24]。ITGAM和ITGAX编码白细胞特异性整合素αM、αX,它们是参与白细胞粘附、迁移,介导巨噬细胞吞噬补体包裹的微粒的作用。CARD9是促进NF-κB通路激活的强促炎分子。2015年,Li等对先前报道的中国队列进行扩展分析,并描述了三个另外的全基因组显著位点,包括ST6GAL1、ACCS和ODF1-KLF10[16],这些基因座可能是亚洲特异性的。2017年,Kiryluk等对2633名欧洲和东亚血统的受试者进行了血清半乳糖缺陷IgA1 (Gd-IgA1)水平的定量GWAS,发现了两个全基因组显著位点,C1GALT1 (rs13226913,P= 3.2×10-11)和C1GALT1C1 (rs5910940,P= 2.7 × 10-8),它们编码IgA1 O糖基化酶促反应所必需的分子伴侣[17]。2021年,我中心[18]在中国大样本人群全面分析了参与中国人群糖基化异常IgA1形成的遗传因素,测定了1127名IgA肾病患者的基因型及其血清中Gd-IgA1水平,共鉴定出2个与Gd-IgA1水平相关的基因座位:GALNT12和C1GALT1,其中GALNT12为新发现,两个基因均参与编码O-糖基化过程的糖基转移酶;且二基因之间存在交互作用,并与IgA肾病的发病风险有关。
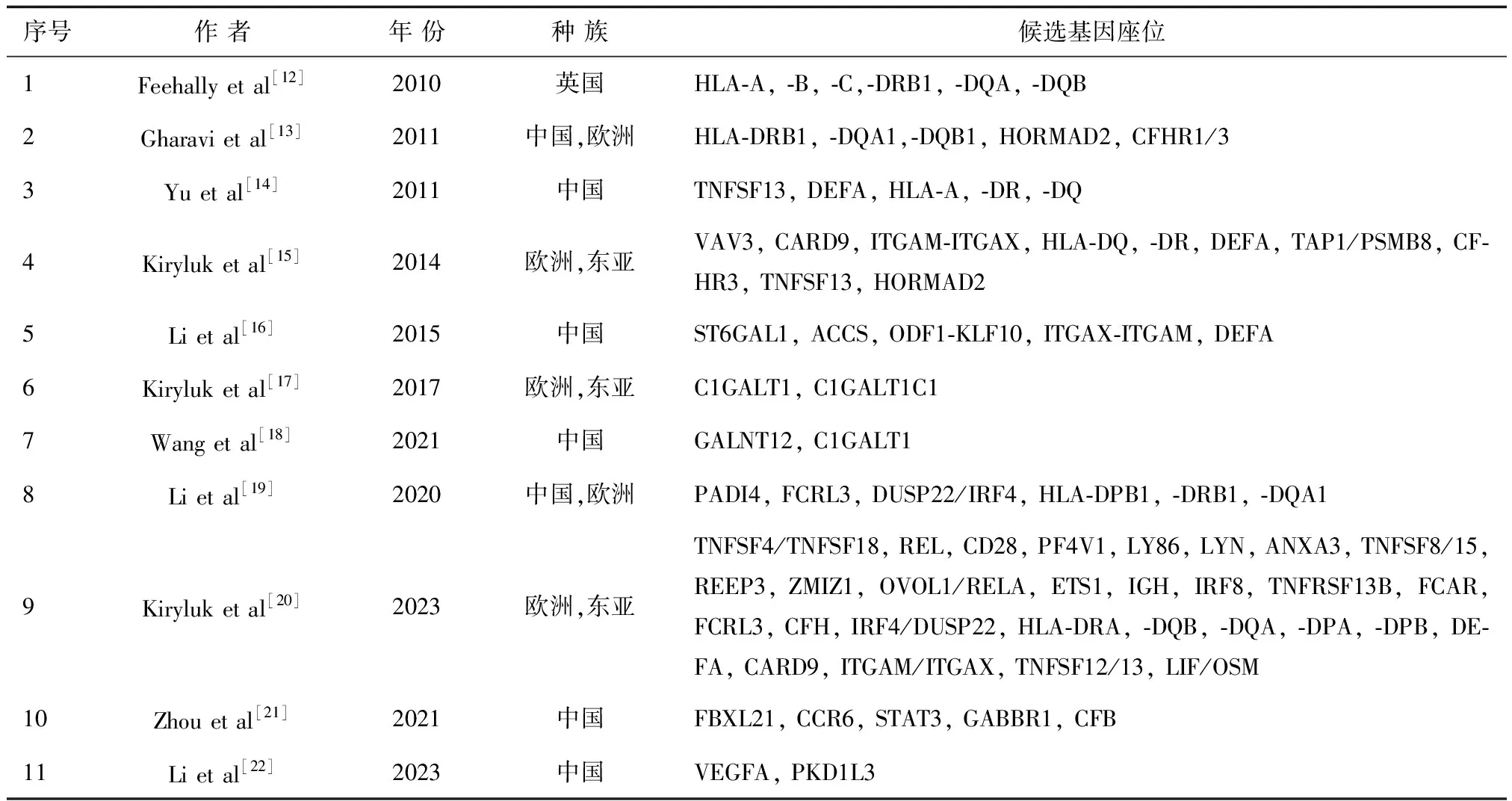
表1 目前GWAS报道的IgAN相关基因座位
近年来,多种族全基因组荟萃分析进一步发现了更多的IgAN的易感相关位点,为阐明IgAN的发病机制和解释不同种族人群之间的易感性差异提供了重要的线索。2020年,Li等[19]通过对 10546 名患者和 21871 名健康对照者进行GWAS的荟萃分析,在1p36.13 (rs2240335)、1q23.1 (rs6427389) 和 6p25.3 (rs6942325) 上发现了三个新的遗传风险位点包括PADI4、FCRL3和DUSP22/IRF4,并且在24个已确认的风险SNPs中显示了7个位点的遗传异质性。2023年,Kiryluk等[20]对 17 个国际队列中的 10146 例 IgAN 病例和 28751 例对照进行了全基因组关联研究和荟萃分析,发现了30个全基因组显著风险位点,解释了11%的疾病风险。其中共有16个新位点,包括TNFSF4/TNFSF18、REL、CD28、PF4V1、LY86、LYN、ANXA3、TNFSF8/TNFSF15、REEP3、ZMIZ1、OVOL1/RELA、ETS1、IGH、IRF8、TNFRSF13B和FCAR。这些研究中新位点的发现为IgAN发病机制的研究提供了新的思路。
3 IgAN易感性的后GWAS和二代测序研究
GWAS研究发现的仅仅是与疾病具有统计相关性的位点,跟生物学关联还存在差距,因为GWAS 所分析的芯片设计基于标签SNP。后续还需根据连锁不平衡信息进行基因型填补或靶向重测序来获得GWAS分析未包含的遗传变异,再利用整合转录组、表观遗传等多组学和基因组结构数据及孟德尔随机化的应用,对相关变异进行基因注释筛选候选的功能位点做进一步的功能注释,接着开展体内外实验来阐明致病变异的分子机制,进而应用于疾病的预防筛查、风险评分和临床治疗等。
然而GWAS固有的局限性包括只能检测到常见的(频率>1%~5%)变异,通常表现出相对较小的效应,且大多位于非编码区域,所以GWAS往往只能解释相对较小比例的疾病遗传度。全外显子组和全基因组测序技术的应用使得我们揭示了更多的非同义编码变体,特别是那些频率较低且具有中等到较大效应的编码变体。
以往大量的GWAS发现22号染色体22q12.2基因座和IgAN有关联,但是这个基因座包含多个基因,包括MTMR3、HORMAD2、LIF和OSM,然而其因果变体和IgAN发病机制的关联原因并不清楚。为了阐明该区域的具体致病变异和致病机制,Wang等[25]基于包含 2762 例 IgAN 患者和 5803 个对照者的 GWAS 数据集对该遗传区域进行精细定位,鉴定了2个与IgAN发病关联的位点:rs4823074和rs16988135,并将rs4823074确定为与永生化淋巴母细胞中 MTMR3 启动子相互作用的候选因果变异。他们进一步用共定位和孟德尔随机化分析发现与血清IgA水平相关的变异在该位点与IgAN风险等位基因共定位的先验概率为88.7%,两者也存在着很强的因果关系。这些结果说明MTMR3/HORMAD2/LIF/OSM 区域的遗传变异能够通过上调MTMR3的表达来升高血清IgA水平,进而影响IgAN发病。此外,体外机制研究表明,MTMR3 依赖于其磷脂酰肌醇 3-磷酸结合域来增加 IgA 的产生;体内功能研究表明 Mtmr3-/-小鼠表现出 Toll 样受体 9(TLR9) 诱导的 IgA 产生和肾小球 IgA 沉积的缺陷。总之,在MTMR3-HORMAD2区域鉴定了2个与IgAN发病关联的位点,并进一步确定其靶基因为MTMR3,阐明了 MTMR3 通过增强 TLR9诱导的 IgA 免疫在 IgAN 发病机制中的作用,表明通过MTMR3抑制靶向黏膜IgA免疫可能代表了一种有前途的IgAN特异性治疗新方法。
包括补体因子 H (CFH) 基因和补体因子 H 相关(CFHR)基因等多个补体调节基因的1q32是IgAN易感性基因座[13]。CFHR5、CFHR2和CFHR1具有二聚化基序,能够形成同源和异二聚体,增强这些蛋白质对C3b的亲和力,使它们能够作为因子H的竞争性拮抗剂发挥作用[26]。为了探索CFHR5变异对IgAN易感性的遗传影响,Zhai等[27]招募了500名IgAN患者和576名健康对照组,对CFHR5的所有外显子及其内含子侧翼区域以及非翻译区域进行了测序,并使用序列核关联检验比较了已识别变异的频率。他们在CFHR5中发现了28个罕见变异,其分布与对照组差异显著。对检测到的28个罕见变异的分析确定了9个具有潜在功能,包括3个改变蛋白质长度的变体和三个通过体外可增加C3b与重组CFHR5结合的变体如CFHR5-M、CFHR5-S和CFHR5-D。这些CFHR5罕见的变异有助于IgAN的遗传风险。2017年,Medjeral-Thomas等[28]发现较高的血清 CFHR-5 水平与IgAN免疫治疗失败、毛细血管内细胞增多以及疾病严重程度的组织学评分(牛津分类 MEST 评分)有关。Zhu等[29]发现高CFHR-5水平增加eGFR下降超过30%或终末期肾病的风险(HR=1.23)。
GWAS的研究形式更容易发现效应较弱的常见变异,而在某种程度上忽略了编码区外显子的罕见突变,后者往往对疾病的影响效应更大。外显子区域在人类基因组中仅占1%,却包含合成蛋白质所需要的重要信息,涵盖了与个体表型相关的大部分功能性变异。随着二代测序技术的发展,研究者们开始关注到罕见变异对复杂疾病性状的影响,但由于测序成本高昂,难以在大规模人群中开展。Illumina公司设计的Human Exome BeadChip外显子基因芯片,包含了一系列已知的少见编码区变异,支持经济高效的大规模基因分型和筛选研究。Zhou等[21]对3363名IgA肾病患者和9879名汉族血统的健康对照者进行了一项基于外显子芯片的两阶段IgAN的关联研究,发现三个非HLA基因区域的FBXL21、CCR6和STAT3和两个HLA基因区域的GABBR1和CFB这5个新的易感基因参与了IgA肾病的发生。Li等[22]对8529例IgAN患者和23224例对照者进行了一项基于外显子组芯片的IgA肾病相关性研究和测序分析,在编码血管内皮生长因子a (VEGFA)的基因中发现了一种罕见的变异VEGFA V167I,这种变异与IgA肾病风险增加两倍显著相关。分子模拟实验表明,该变异可能引起构象改变,使VEGFA蛋白更加稳定和紧凑以及增加与其受体VEGFR1和VEGFR2的结合亲和力。然而这些发现的蛋白编码变异需要进一步的功能实验来确定其IgA肾病发病机制中的作用。
4 IgAN GWAS的临床转化
4.1 疾病风险预测IgAN是复杂的多基因疾病,如前所述,GWAS已为IgAN积累了丰富的疾病相关基因变异位点。全基因组多基因风险评分(genome-wide polygenic risk score,GPS)的计算是风险等位基因的加权总分,可用于识别疾病高危个体以进行临床干预,并提供比传统临床风险评分更多的信息有望开展分层筛查。
Sukcharoen等[30]用从最大的欧洲GWAS中提取的 14 个SNP 生成IgA 肾病遗传风险评分 (IgAN-GRS),并在来自英国肾小球肾炎DNA银行(UKGDB)的464例经活检证实的IgAN欧洲病例和379767名UKBB欧洲人中进行验证,并使用IgAN-GRS的平均值来计算来自379767名UKBB欧洲人的14181名血尿和潜在IgAN的比例。结果显示UKBB 人群中 15%~25%的不明血尿可能患有 IgAN,而该群体通常可能不会接受肾活检或诊断。此研究表明多基因风险的非侵入性使用可能可以识别大量人群中肾脏表型的可能病因。近期Kiryluk等[20]设计并优化了 IgAN 的 GPS,并在独立的德国慢性肾脏病 (GCKD) 研究中进行测试时,解释了 7.3% 的疾病风险(P= 3.1 × 10-12;C-统计量=0.65;95%CI=0.61~0.68)。在2879名长期随访的IgAN患者中,高GPS患者有更高的肾衰风险[HR=1.17(95%CI:1.09~1.24),P=3.3×10-6]。位于GPS20%高分的个体发生尿毒症的风险增加34%[HR=1.34 (95%CI:1.15~1.56),P=2.0×10-4],而在前10%的个体发生尿毒症的风险增加48%[HR=1.48 (95%CI:1.22~1.79),P=6.6×10-5]。表明较高的多基因评分不仅增加疾病易感性,也增加进展至尿毒症的风险。未来的研究需要关注多基因分层是否有助于诊断、临床风险评估或治疗反应预测。
4.2 药物开发IgAN的GWAS研究的结果可概括为几种关键的生物学途径,为临床新药的研发提供了理论基础。一项研究[20]对IgAN蛋白质互作分析中发现富集在趋化和细胞因子通路和细胞因子配体-受体交互作用,包括APRIL和它的受体TACI,IL-6相关细胞因子-受体对(IL6-IL6ST、LIF-LIFR/IL6ST和OSM-OSMR/LIFR/IL6ST)。值得关注的是,已知APRIL会改变IgA1的糖基化,IL6、LIF和OSM参与粘膜免疫,IL-6和LIF可促进致病性Gd-IgA1的产生。这些细胞因子有望开发未来的生物治疗。同时对候选基因进行了药物靶点的分析时,在前 26 个高度优先的“生物候选药物”中,总共有13个基因座编码的17个蛋白质已有药物,11个基因座编码的14个蛋白质预测可成药。这些靶点中有11个(42%)是现有药物。其中包括以下内容:①目前正在进行肾小球疾病临床试验的补体旁路途径抑制剂如APL-2;②通过抑制 APRIL 或 TACI 来阻断B细胞激活的药物如阿塞西普;③通过靶向CD28抑制T细胞活化的药物,如贝拉西普(批准用于同种异体移植排斥)或阿巴西普(批准用于类风湿性关节炎);④抑制 IL8 (ABX-IL8) 或 IL8 受体的药物如克霉唑;⑤抑制 NF-κB 通路的药物,例如已经处于肾小球疾病临床试验中的巴多索隆。在IgAN中一些致病基因(如 CARD9、ITGAX、PF4V1、CFHR1 或 FCAR)尚无有效的药物抑制剂。部分基因编码具有保护性的分泌蛋白,例如 FCRL3 和 TNFSF4,这表明针对它们的上调可能会提供合理的治疗策略。这些研究发现了重要的疾病靶标,从而为开发出特异性更强的新药指明方向,同时为不同疾病相同信号通路中的老药新用提供依据。但是,仍然需要进行机制研究首先确认靶基因的正确性和致病机制。
5 总结和展望
继 2010年第一个IgAN的GWAS报道之后,随着GWAS的样本量的增加和功能性变异研究的开展,大量的与IgAN相关的易感性位点和其生物学功能逐渐被揭示。未来需要整合遗传、临床和生物信息学数据集的工作来进一步阐明IgAN的发病机制。首先,全面检测遗传变异对于揭示 IgAN 的发病机制至关重要。迄今为止发现的风险位点解释了11%的疾病风险。通过对更大的样本量进行GWAS,使用基于插补的荟萃分析,对IgAN和自身免疫或炎症相关疾病进行交叉表型遗传分析,可能有望全面揭示其遗传背景。不同于GWAS鉴定的位点多为常见的非编码区变异,全外显子测序和全基因组测序可以来识别罕见变异,可能解释复杂性状中遗传力缺失的部分。其次,对于已确定的位点,使用生物信息学方法解释其功能非常重要,包括公共数据库的计算机分析,通路分析和功能验证。第三,也许最重要的顾虑是IgAN的特征是显著异质性的病理和临床表现,需要有IgAN更精细的表型表征,以帮助预测治疗和预后意义。所以未来GWAS 的研究是对更精细表型的生物学阐释,将遗传学与生物学联系起来,开发基于遗传信息的药物治疗,改进临床风险预测,并确保这些对疾病产生积极影响。
