大豆油中蛋白质残留量测定方法学研究
2024-01-18巫建新唐顺之魏劭恒杨玉琼李佳俐许文东
◎ 巫建新,唐顺之,李 遥,白 柏,魏劭恒,杨玉琼,李佳俐,许文东
(1.广州白云山汉方现代药业有限公司,广东 广州 510240;2.中药制药过程技术与新药创制国家工程研究中心,广东 广州 510240;3.广东省药用脂质重点实验室,广东 广州 510240)
大豆油是日常生活中常见的一种植物油,主要成分为脂肪,是人体重要的营养来源,可以为机体提供能量,维持机体体温,支撑脏器,促进糖代谢和脂溶性维生素吸收等作用[1]。大豆油含有丰富的脂肪酸,其中亚油酸含量在48%~58%,亚油酸有助于降低血清胆固醇和抑制动脉血栓的形成[2],具有极高的营养价值。
通过压榨或者溶剂提取得到的大豆毛油含有较多的杂质,不能直接食用,必须经过脱胶、脱酸、脱色、脱臭等精炼工序处理,去除其中残留的胶质、色素、氧化物、磷脂、蛋白质、游离脂肪酸、金属元素等大部分杂质,才能有较好的外观、气味、滋味和贮藏稳定性。其中油脂和蛋白质混合受热会产生更多的苯并芘[3],而苯并芘是一种强致癌物[4],我国食用植物油卫生标准规定食用植物油中苯并芘的限量不超过10 μg·kg-1。因此控制大豆油中蛋白质残留量有利于减少大豆油煎炸过程中苯并芘的产生,提高食用大豆油的安全性。
蛋白质测定方法有定氮法、福林酚法、双缩脲法、燃烧法、考马斯亮蓝法以及紫外-可见分光光度法等[5]。双缩脲法灵敏度偏低,考马斯亮蓝法对于不同样品的蛋白质测定结果可能存在一定偏差,紫外-可见分光光度法易受溶液中的杂质影响,标准曲线难以准确绘制。此外由于大豆油不溶于水,无法通过溶解后显色测定蛋白质;而采用定氮法,可以对大豆油进行消解,去除大豆油基质的影响,测定氮含量,再换算为蛋白质含量,更适合测定大豆油中的蛋白质残留量。本文旨在通过开发适用于大豆油中蛋白质残留量的分析方法,测定大豆油中蛋白质残留量,为评估大豆油中蛋白质残留风险提供依据。
1 材料与方法
1.1 材料
硫酸(AR)、乙醇(AR)、硼酸(AR)、氢氧化钠(AR)、甲基红指示剂、溴甲酚绿指示剂,以上试剂均购自广州化学试剂厂;盐酸滴定液(0.02 mol·L-1),购自深圳市博林达科技有限公司;硫酸铵(≥99.99%),购自上海阿拉丁生化科技股份有限公司。
1.2 仪器与设备
Kjeltec® 8420 自动凯氏定氮仪,丹麦福斯有限公司;MS204S 电子天平,梅特勒-托利多国际有限公司;消化炉,丹麦福斯有限公司。
1.3 试验方法
1.3.1 溶液配制
硼酸(H3HO3)溶液(10 mg·mL-1):称取10 g H3HO3,用水溶解并稀释至1 000 mL;氢氧化钠(NaOH)溶液(400 mg·mL-1):称取40 g NaOH 加水溶解后,放冷,并稀释至100 mL;甲基红乙醇溶液(1 mg·mL-1):称取0.1 g 甲基红,用乙醇溶解并稀释至100 mL,可用超声水浴促进溶解;溴甲酚绿乙醇溶液(1 mg·mL-1):称取0.1 g 溴甲酚绿,用乙醇溶解并稀释至100 mL;混合指示液:每10 L 硼酸溶液添加70 mL 甲基红乙醇溶液与100 mL 溴甲酚绿乙醇溶液制成;硫酸铵[(NH4)2SO4]标准溶液:精密称取105 ℃干燥2 h 的(NH4)2SO4约33 mg,置于100 mL 容量瓶中,用水溶解并稀释至刻度,摇匀,即得每1 mL 含(NH4)2SO4约0.33 mg 的硫酸铵标准溶液。
浓度梯度基准物质溶液:分别精密吸取硫酸铵标准溶液0.2 mL、0.4 mL、0.8 mL、1.2 mL、1.6 mL、2.0 mL 于干燥的250 mL 凯氏消化管中,按照样品制备方法操作制得系列浓度梯度基准物质溶液。
1.3.2 样品制备
取供试品0.5 g,精密称定,移入干燥的250 mL凯氏消化管中,加入2 片凯尔特催化片Cu 3.5 及12 mL硫酸,摇匀。待以上样品完全浸湿后移至预热好的FOSS 消化炉(420 ℃)中,套上排废罩,开启冷凝水(开始时需将水抽气泵全开,5 min 后调小抽气泵水流使酸雾恰好被吸入涤气罩),开始消化,直至全部样品变为透明的蓝绿色澄清液体(大约90 min)后,消化完毕,将消化管连同消化管架和涤气罩一起从消化炉中取出,冷却10~20 min 后,加入50 mL 水,备用。同时制备样品空白溶液。按照 Foss Kjeltec® 8420 自动分析定氮仪的要求,设定凯氏定氮分析程序进行测定。
1.3.3 试样中蛋白质的计算
计算公式为
式中:X为试样中蛋白质的含量,g/100 g;v为样品溶液消耗盐酸滴定液(0.02 mol·L-1)的体积,mL;v0为试剂空白消耗盐酸滴定液(0.02 mol·L-1)的体积,mL;c为盐酸滴定液浓度,mol·L-1;0.014 0 为盐酸滴定液(0.02 mol·L-1)相当的氮的质量,g;m为供试品的质量,g;6.25 为供试品中氮换算为蛋白质的系数。
2 结果与分析
2.1 标准曲线
使用基准硫酸铵制备6 个浓度基准物质溶液并使用自动分析定氮仪测定,以氮含量为横坐标,滴定体积为纵坐标,绘制标准曲线,得出回归方程和相关系数。如表1 所示,氮含量在0.063 6~0.635 9 mg(即相当于样品中蛋白质含量在0.080%~0.795%)时线性关系良好。

表1 线性和范围表
2.2 取样量考察
分别称取同一大豆油供试品0.5 g、1.0 g、1.5 g、2.0 g、5.0 g,按照供试品溶液的制备方法平行制备2 份。结果显示,称样量为1.0 g、1.5 g、2.0 g、5.0 g 时,供试品未能完全消解,消解物呈黑色团块状,并膨胀至消解管上部,硫酸也因长时间消解而挥干。称样量为0.5 g 时,供试品可消解为蓝绿色透明状液体,因此方法采用的称样量为0.5 g。
2.3 检出限和定量限
制备7 份样品空白溶液并测定,通过空白试验标准偏差计算方法检出限(Method Detection Limit,MDL),计算公式如下
式中:n为样品的平行测定次数;t为自由度为n-1,置信度为99%时的t分布值(单侧),此处t=3.143;S为n次平行测定的标准偏差。
计算结果如表2 所示,该方法的检出限为0.027%。

表2 检出限结果表
分别称取同一大豆油供试品9 份,每3 份各添加硫酸铵标准溶液0.05 mL、0.08 mL、0.10 mL,按照供试品溶液制备方法,制备理论氮加入量为0.035 mg、0.056 mg、0.070 mg 的加标供试品溶液并进行测定。结果如表3 所示,当氮加入量为0.070 mg 时,加标回收率在92.7%~98.3%,重复性(n=3)RSD ≤4%,符合定量限要求,此时对应的含量0.087%即为方法定量限。

表3 定量限结果表
2.4 精密度
分别称取同一大豆油供试品6 份,添加硫酸铵标准溶液0.10 mL,按照供试品溶液制备方法,制备理论氮加入量为0.070 mg的加标供试品溶液并进行测定,计算6 份样品氮含量的RSD 值作为重复性结果。另一人员同法操作,计算两人员12 份样品氮含量的RSD值作为中间精密度结果。同一人员连续3 d 同法操作,计算18份样品氮含量的RSD值作为日间精密度结果。如表4 所示,重复性(n=6)、中间精密度(n=12)和日间精密度(n=18)的RSD ≤4%,说明本方法精密度良好。

表4 精密度结果表
2.5 准确度
分别称取同一大豆油供试品6 份,添加硫酸铵标准溶液0.10 mL,按照供试品溶液制备方法,制备理论氮加入量为0.070 mg 的加标供试品溶液并进行测定,计算加标回收率。结果如表5 所示,加标回收率在92.1%~97.9%,重复性(n=6)RSD ≤4%,符合方法学要求,说明本方法准确度良好。
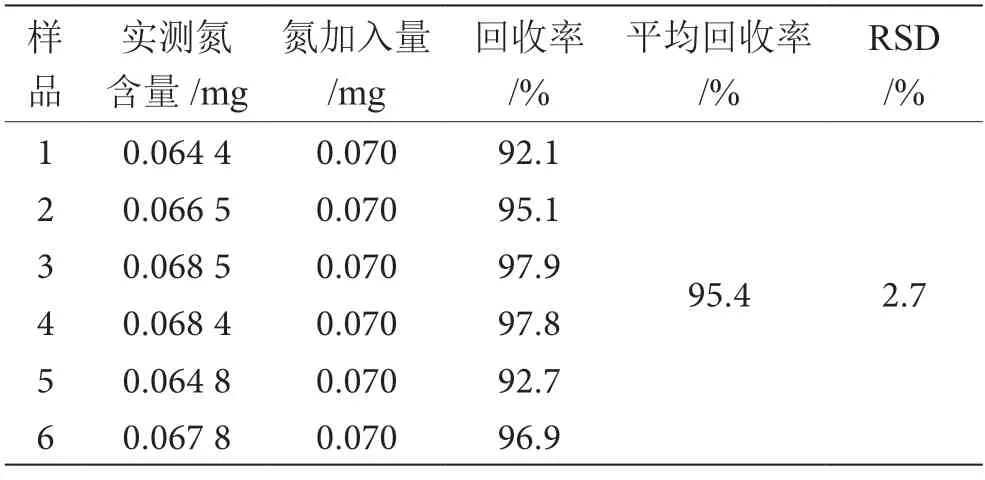
表5 准确度结果表
2.6 样品测定
取多批白云山汉方公司自制的大豆油样品按验证后的蛋白质残留量测定方法进行检测,结果显示大豆油中蛋白质残留量均低于检出限(0.027%),说明大豆油中蛋白质残留风险较低。
3 结论
本文对氮测定法进行方法验证,开发出适用于大豆油蛋白质残留量测定方法,该方法最高取样量是0.5 g,继续增加取样量会导致消化不完全,影响检测进行。该方法检出限和定量限分别为0.027%和0.087%。本方法在0.080%~0.795%线性关系良好,且精密度和准确度均符合方法学要求,能满足大豆油蛋白质残留量测定,可以为大豆油蛋白质残留的安全性评估提供方法依据。
