基于机器学习的蛋白质编码区识别
2024-01-13包晓娜何黎黎崔景安
包晓娜,何黎黎,崔景安
(北京建筑大学 理学院 北京102616)
大多数真核生物的编码区是不连续的,编码蛋白质的序列在基因序列中被非编码序列隔开(见图1)。编码的序列又称为外显子(Exon),携带着遗传信息,能够决定和指导生物的性状;非编码序列又称为内含子(Intron)[1]。如果一个基因有n个内含子,一般总是把基因的外显子分隔成n+1个部分。且内含子的核苷酸数量比外显子多许多倍[1-2]。因此,外显子和内含子的准确识别是一个具有挑战性的研究。外显子和内含子区分也有助于研究基因功能、基因表达、基因注释、基因转录调控,对于内含子功能的研究也具有一定的辅助作用[3-4],故外显子和内含子的分类具有重要的意义。
多年来,学者们已经提出了基因编码区(外显子)预测的多种方法。一般可以分为基于同源比对的方法和不依赖同源比对的方法。基于序列同源性的方法是以现有的基因数据库为标准,对待检测DNA序列进行相似性识别,从而根据已有经验判断未知序列的外显子和内含子区域。BLAST[5]、MUSCLE[6]是常见的比对工具,近年来也有诸如GeMoMa[7]的基因预测程序被提出。基于序列同源性的方法准确率较高,但测序成本高、比对效率等因素制约了该项技术的发展。基于此,许多的学者将研究重点转向不依赖比对技术的模型。数字信号处理技术在该领域发挥着关键的作用[8]。且数字信号处理前通常需对DNA序列进行数值映射[9]。VOSS[10]是一种广泛使用的固定映射技术,它将DNA序列转化为4个二进制指示符序列XA[n],XC[n],XG[n],XT[n]。核苷酸在特定碱基位置出现用1表示,未出现用0表示。Z曲线理论[11]是基于物理化学性质的映射方式。利用传统四面体的对称性开发,它将DNA或RNA序列映射到折叠曲线中。Z曲线表示出DNA序列携带的所有信息[8],可用于基因鉴定和DNA或RNA序列分析[12]、识别细菌和古细菌基因组中蛋白质编码基因[13]等。此外,在众多序列编码方法中,k字符相对频率技术(k-mer)[14]是较常见和简便的方法。图2展示了当k为4步长为1时的短序列的k-mer生成过程。机器学习的迅猛发展也为蛋白质编码区的识别带来了许多新的算法。如CNN-MGP[15]、GeneMark EP+[16]、DBN[17]。CNN-MGP[15]是用于宏基因组学基因预测的卷积神经网络,能够提取编码区和非编码区的特征。GeneMark EP+[16]是用于真核基因预测的算法和工具。深度置信网络DBN[17]通过多层玻尔兹曼机对DNA序列进行数值转换,用深度置信网络模型对外显子和内含子分类判别。尽管已经有许多的外显子与内含子分类方法被提出,但是准确率、敏感度、特异度、AUC值等评价参数还有待提升。
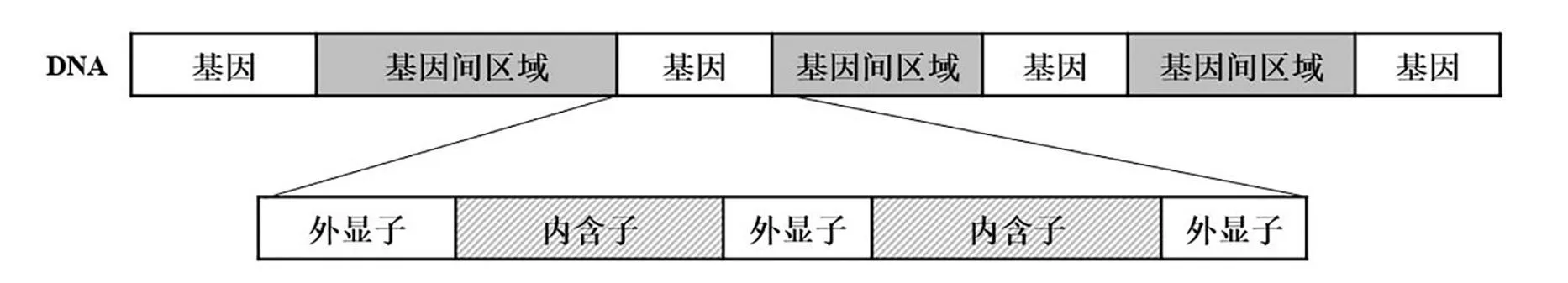
图1 真核生物外显子与内含子交替示意图Fig.1 Schematic diagram of exon intron alternation in eukaryotic coding region

图2 k字符相对频率技术提取k-mer示意图(k=4)Fig.2 Schematic diagram of k-mer extraction by k-character relative frequency technology(k=4)
将数值映射和机器学习分类器相结合,提出了一个组合算法(具体流程见图3)。首先,给定一个外显子或内含子,将其通过密码子与氨基酸的对应转换为特定的氨基酸序列,此处的转换不同于标准的翻译过程。然后,利用经典的k-mer技术获取序列的特征向量。最后,将外显子与内含子的特征向量输入逻辑回归分类器中,训练模型并识别蛋白质编码区(外显子)。利用真核生物基准数据集HMR195和BG570对模型进行了五折交叉验证,AUC值分别达到了0.981 3和0.987 4。将两个数据集合并计算时,敏感度和特异度分别为0.954 1、0.942 8。通过对比发现,新算法的识别结果明显优于VOSSDFT、传统的贝叶斯判别等方法。新算法识别HMR195和BG570数据集的时间为1.46 s、3.58 s,表明组合模型能够高效又准确地鉴定真核生物的外显子和内含子。
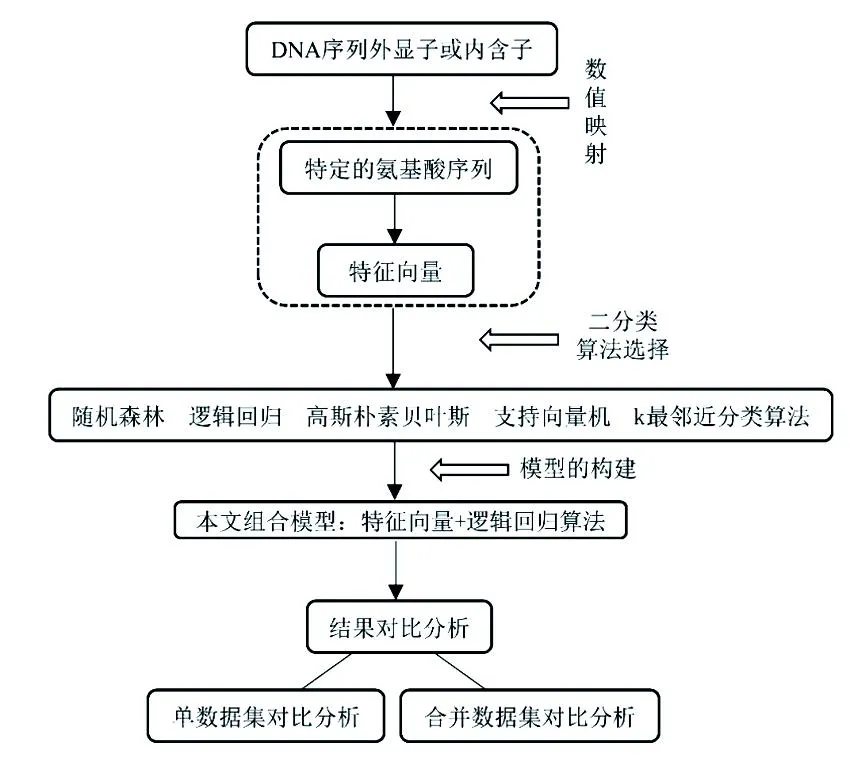
图3 本文算法的框图Fig.3 Block diagram of the algorithm in this article
1 数据
1.1 数据的获取
本文对真核生物的DNA序列进行编码区判别分析,实验中用到2个基准数据集,分别是HMR195[18]和BG570[19]数据。HMR195数据由195个哺乳动物DNA序列组成,包括人类、小鼠和大鼠,共948个外显子。BG570是指由570个脊椎动物序列组成的基因组测试数据集,共2 649个外显子。两个数据集可从网址http://www.imtech.res.in/raghava/genebench中获取。基准数据集的长度范围、外显子数目和内含子数目如表1所示。为了保证对外显子和内含子分类的全面性,将短的(长度低于20 bp)外显子和内含子序列也加入了实验中。

表1 基准数据的外显子和内含子分布表Table 1 Exon and intron distribution table ofbenchmark data
1.2 数据的预处理
1.2.1 DNA序列的数值转化
在实现外显子和内含子的精准分类与预测前,通常需要对DNA序列进行数值映射,即将DNA序列转化为一个数值形式的表示[17]。本文提出了一个全新的DNA序列数值化映射方法,结合k-mer技术[14],将DNA序列中的外显子和内含子分别转化为一个特征向量。下面介绍特征向量的提取过程:
给定一个外显子ACAGCGACC:第1步,从第一个核苷酸A处开始,通过每次仅移动一个核苷酸,将外显子转化为一段特定氨基酸序列,具体为,‘ACA’对应氨基酸T,‘CAG’对应氨基酸Q,‘AGC’对应氨基酸S,‘GCG’对应氨基酸A,‘CGA’对应氨基酸R,‘GAC’对应氨基酸D,‘ACC’对应氨基酸T,由此得到一段特定氨基酸序列为TQSARDT;第2步,结合经典的k字符相对频率技术,规定k值从1至5变化,将TQSARDT转化为特征向量。假设k=2,则2-mer种类包括TQ、QS、SA、AR、RD、DT。特征向量由2-mer频数构成,即(fTQ,fQS,fSA,fAR,fRD,fDT)=(1,1,1,1,1,1),其中fTQ表示TQ的频数。
1.2.2 DNA序列的特征提取
DNA序列特征提取源于特定氨基酸序列k-mer的种类和数值频率。具体来说,特征向量的元素(即所有的k-mer种类)是DNA序列的特征,即1.2.1节提到的TQ、QS等。通常来讲,一段氨基酸序列中的k-mer种类数为20k。但是,由于特定氨基酸序列的转化不同于生物学中标准的翻译过程,且存在不同的密码子对应同一种氨基酸,所以本算法的k-mer种类数远远少于20k,这大大节约了计算时的内存消耗。以脯氨酸P为例,如图4,它由4个密码子编译CCT、CCC、CCA、CCG,由本文的转化过程,P后的下一个氨基酸共5种,分别为亮氨酸L、组氨酸H、谷氨酰胺Q、脯氨酸P、精氨酸R,远少于20种。因此,每个氨基酸后可能出现的氨基酸种类少于20种。最终k-mer组合种类数随之大大减少,也就是说本文的转化过程大大降低了特征向量的维度。表2列出了k=2时的全部95种特征向量的元素特征。
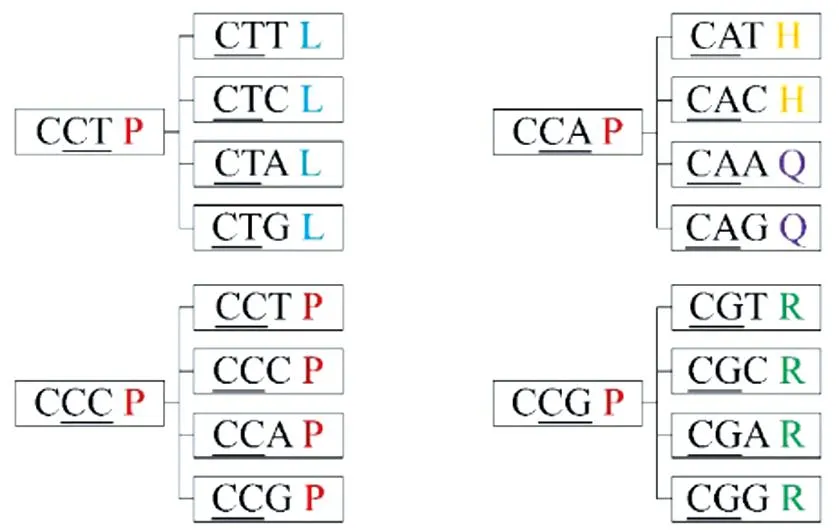
图4 脯氨酸P后面会出现的氨基酸种类示意图Fig.4 Schematic diagram of amino acid types that will appear after P

表2 k=2时,特征向量的95种元素Table 2 When k = 2, 95 elements of eigenvector
表3列出了部分短外显子或内含子的特征向量(以k=2为例)详细求解过程。规定外显子类别为1,内含子类别为0。
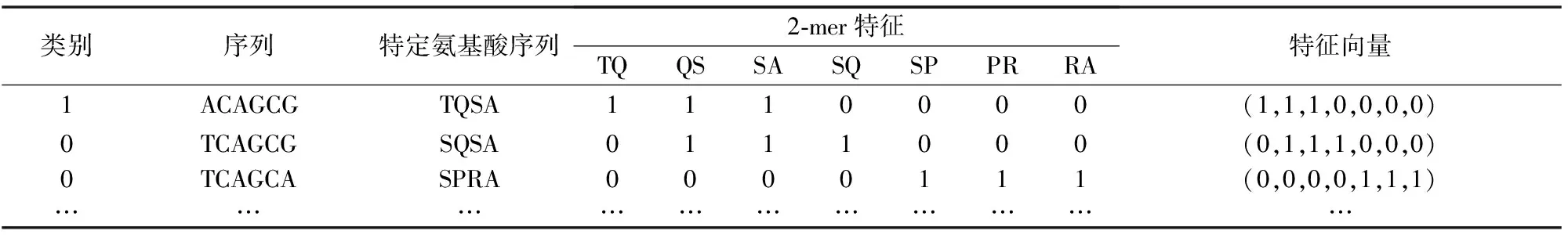
表3 当k=2时,部分外显子和内含子序列的特征向量Table 3 When k=2, eigenvectors of some exon and intron sequences
2 模型的构建
2.1 二分类算法的选择
在完成DNA序列的数值转换后,为了找到最适合特征向量的二分类模型,本文对五种分类器进行了尝试和验证,分别是随机森林(Random forest)[20]、逻辑回归(Logistic regression)[21]、高斯朴素贝叶斯(Gaussian naive bayes)[22]、支持向量机(SVM)[23]、k最邻近分类算法(KNN)[24]。计算时,采用五折交叉验证[25]。五折交叉验证是判断分类器性能的一种统计分析方法。它将原始数据分为5组,不重复地抽取其中4组作为训练集,剩余的1组作为测试集,共得到5种测试结果,最终取用平均数。
为了对5种不同的算法进行有效的对比和测度,此处使用三个评价指标ROC(Receiver operating characteristic)曲线、AUC值和近似相关系数AC值。ROC曲线[26]是以假阳率(False positive rate)作为横轴线(成本),以真阳率(True positive rate)作为纵轴线(收益),来说明在各种阈值条件下的假阳率和真阳率的关系曲线。ROC曲线与对角线的距离愈接近,表明试验中识别编码区与非编码区的能力愈弱,亦即该方法的分类预测能力愈弱。为了更准确地描述算法的判别能力,通常将ROC曲线下的区域面积用AUC[26]进行定量和比较,AUC数值愈接近1,说明分类的有效性越好。近似相关系数AC[26]是一种得到普遍认可的综合评估指标,TP(True positive)为外显子被正确预测的个数,FP(False positive)为预测为外显子但实际为内含子的个数,TN(True negative)为内含子被正确预测的个数,FN(False negative)为预测为内含子但实际为外显子的个数。此外,为了检验结果的统计学显著性,采用Delong检验[27]对ROC-AUC进行成对比较,p<0.05被认为具有统计学意义。
(1)
具体实验结果如图5、图6和表4所示。图5中,k=2时,在HMR195数据集,逻辑回归的AUC平均数分别为0.981 3,明显高于其他模型的结果。如图6,BG570数据集也得到类似的结果,逻辑回归算法在所有k值优于其他算法。

图5 HMR195数据集中,五种算法AUC值的热图Fig.5 In HMR195 data set, heatmaps of AUC values of five algorithms
AC值对比结果如表4所示,k=2时,逻辑回归取得了最大的AC值。AC值兼顾了TP、TN、FP、FN四个参数的值。AC值越大,表明分类效果越好。同时可以发现,当k取其他值时,逻辑回归算法相较其余四种方法也具有明显的优势。因此,由特征向量与逻辑回归组合的分类模型较准确。
2.2 组合模型的确定
最终,组合模型确定为特征向量与逻辑回归分类器的结合。首先,将DNA序列转化为特定的氨基酸序列;其次,由特定氨基酸序列得到特征向量。最后,将特征向量放入逻辑回归分类器中,获得外显子和内含子的预测结果。
如图7,选取五折交叉验证中的一次实验结果,画出ROC曲线图(k=2)。可以明显看出,组合模型最贴近面积为1的四边形线,分类效果较好。并且,HMR195的结果具有统计学显著性(逻辑回归VS随机森林:p=5.07×10-8;逻辑回归VS朴素贝叶斯:p=4.99×10-16;逻辑回归VS支持向量机:p=7.74×10-10;逻辑回归VS k最邻近算法:p=8.91×10-7)。BG570数据的试验结果也显著(逻辑回归VS随机森林:p=8.05×10-16;逻辑回归VS朴素贝叶斯:p=3.70×10-54;逻辑回归VS支持向量机:p=4.67×10-9;逻辑回归VS k最邻近算法:p=1.24×10-7)。
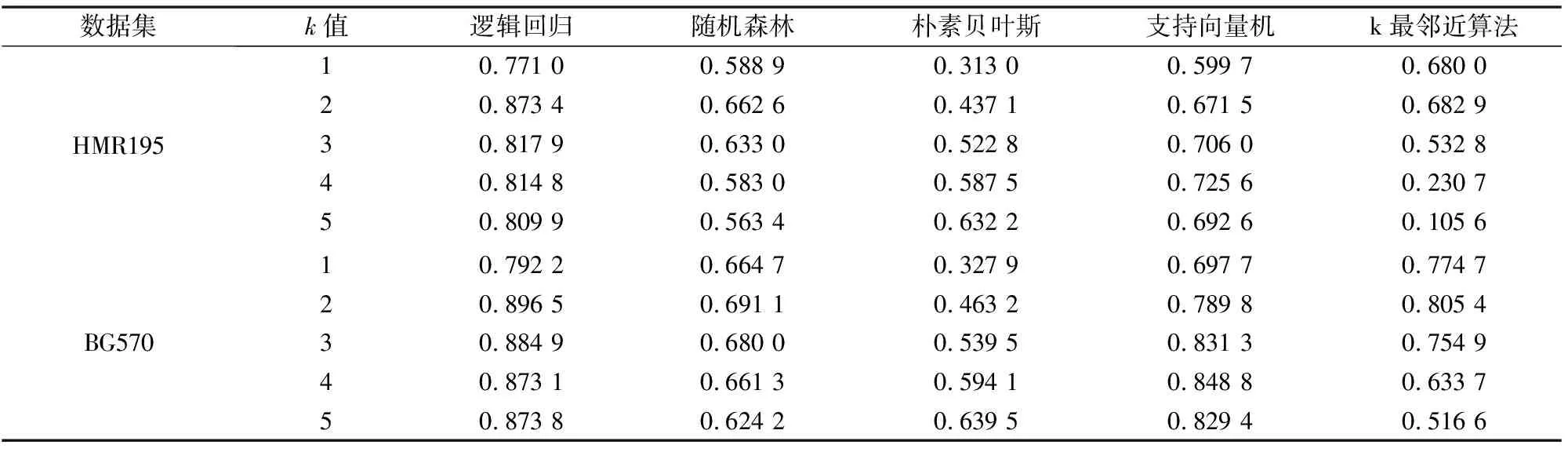
表4 k从1至5,5种算法的AC平均值Table 4 K from 1 to 5, mean AC value of 5 algorithms
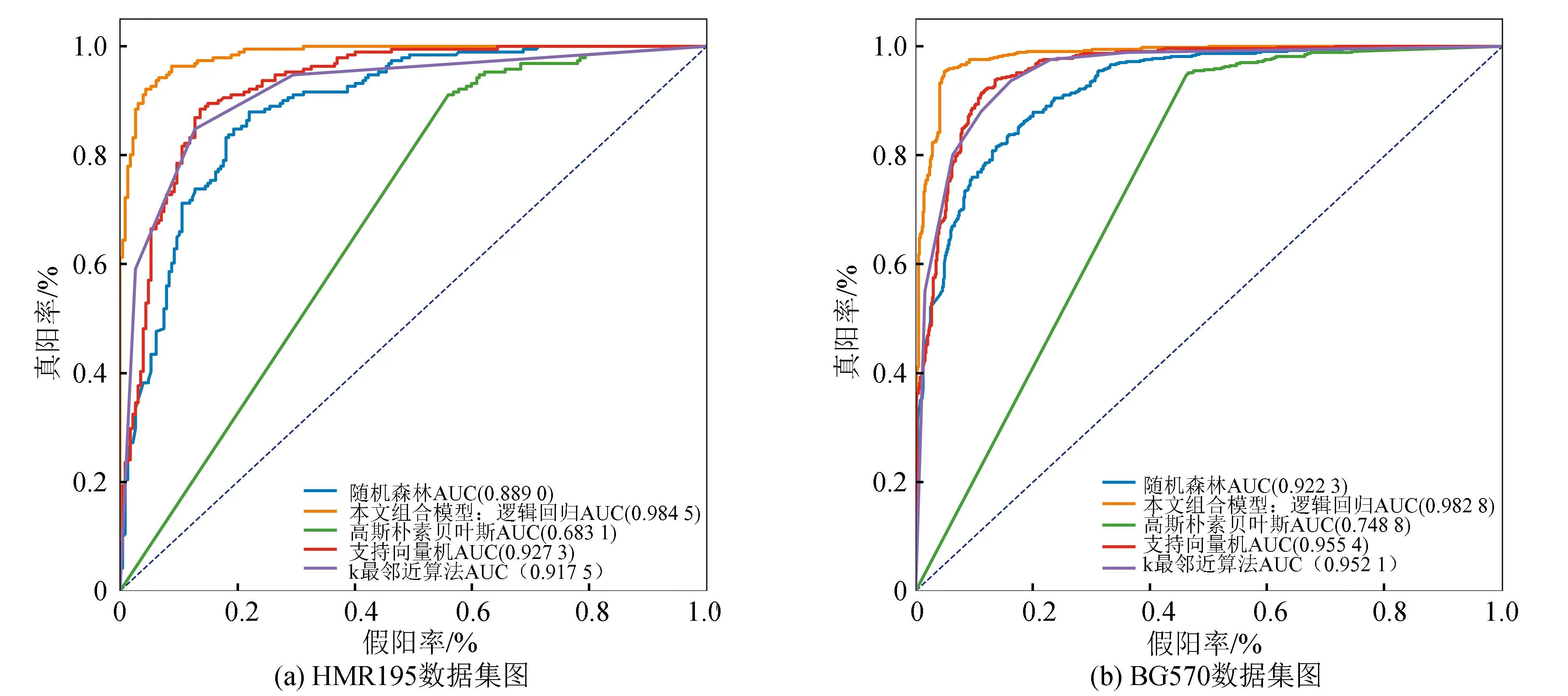
图7 五个算法模型的ROC曲线图Fig.7 ROC curves of 5 algorithm models
3 实验结果
3.1 单独数据集对比分析
为了说明本文新方法与其余方法的优劣,将其与经典的VOSSDFT[10,28]、EIIPDFT[28-29]、SPDFT[28,30]和Code13-Marple[28]进行了比较。VOSSDFT、EIIPDFT、SPDFT均是基于离散傅里叶变换的技术(Discrete Fourier Transform, DFT)来区分真核生物外显子和内含子[10,29-30]。Code13-Marple是一种基于自回归谱分析和小波变换的集成算法。由表5,以HMR195为例,新方法(k=2)的AUC值达到了0.981 3,比其余四种方法分别高出了0.418 7、0.470 0、0.385 1、0.263 4;在BG570数据集上,AUC和AC值也远远超过其余四种模型中的最大值。新算法明显优于其他三种传统的基于DFT的方法和Code13-Marple。
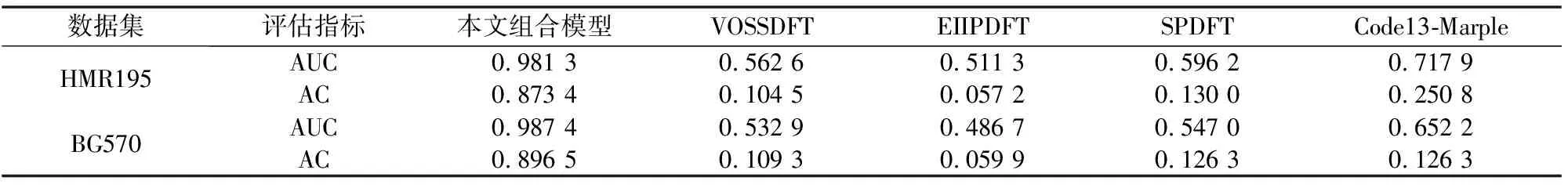
表5 组合模型与其他方法的比较Table 5 Comparison of eigenvector method with other methods
3.2 合并数据集对比分析
为验证算法在较大数据集上的分类效果,将HMR195和BG570两组数据合并得到合并数据集,共3 597个外显子、4 354个内含子。此外,为了更加全面的评估组合模型的性能,增加了准确率、敏感度、特异度以及运行时间这四个对比维度,并与经典的贝叶斯判别法[31]进行比较。贝叶斯判别法是进行判别分析的一种多元统计分析方法。合并数据集后,k值取3时本文算法得到最好的预测结果。
(2)
(3)
(4)
表6是两种方法的对比分析表,其中准确率acc[26]为全部序列中被正确预测的序列的比例;敏感度Sn[26]为所有实际外显子中被正确预测为外显子的比例;特异度Sp[26]为所有真实的内含子被正确预测为内含子的比例。在合并后的较大数据集上,组合模型的敏感度Sn为0.954 1远远大于贝叶斯判别法的0.787 2。在运行时间方面,组合模型只需要8.91 s,而贝叶斯判别法需要27.28 s。因此,本文方法不仅适用于小数据集,在较大数据集上同样表现优异,并且运行速度快于贝叶斯判别法。本文组合模型以及贝叶斯判别法的计算基于处理器为Intel(R) Core(TM) i7-8550U CPU@1.80 GHz和16.0 GB RAM的设备,使用Python3.8编程获得。
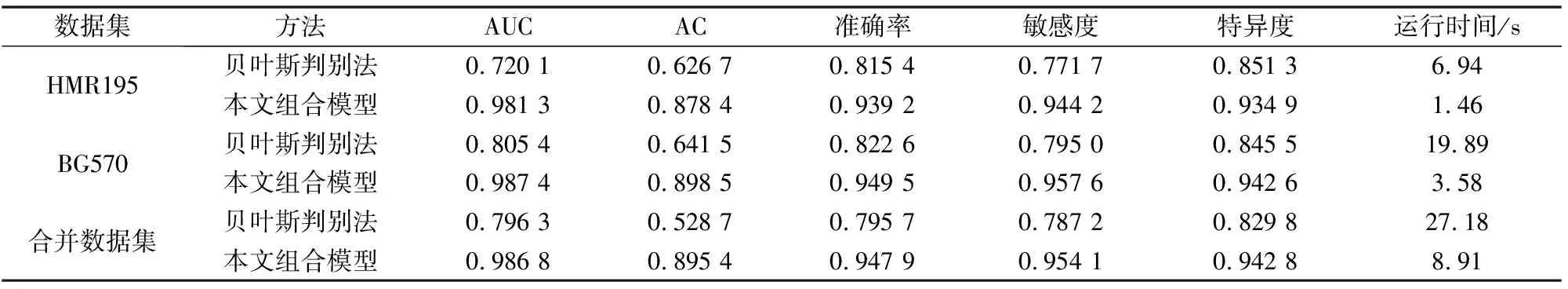
表6 二种模型的比对结果分析表Table 6 Analysis table of comparison results of two models
4 结论及展望
本研究提出了一个基于特征向量的数值映射方法,之后结合逻辑回归算法,对基因外显子和内含子实现了精确的分类。将组合模型运用到编码区识别,给出了一个全新的研究视角。为了证明组合模型的可行性,利用HMR195和BG570两个真核生物数据集,将其与现有的成熟方法进行了对比(见表5和表6),均证明了它的有效性。此外,为证实模型在更大数据集上的效果,本文新收集了462条人类DNA序列[32]进行试验,共包含2 843个外显子,2 381个内含子。全部数据可从网址https://www.fruitfly.org/sequence/human-datasets.html获取。当全部数据共同训练时,共6 440个外显子,6 735个内含子。本文方法实验结果:acc、Sn、Sp、AC、AUC的值分别为0.957 7、0.966 6、0.949 0、0.915 5、0.989 4(k=2)。当扩大数据集后,组合模型对于外显子和内含子依然能起到很好的识别效果。其次,1.2.2节中特征向量的提取过程充分利用了密码子的简并性,降低了特征向量的维度。然而本文还未将外显子和内含子的结构信息作为特征的重要因素,之后的研究中会考虑加入结构信息,从而进一步提升模型的性能。并且,本文后续研究仍将扩大样本量,尝试更多更全面物种的蛋白质编码区分类,争取构建快速便捷的外显子与内含子识别工具。
