基于多层基因网络的关键基因识别算法
2024-01-13魏丕静刘晶晶赵永敏苏延森郑春厚
魏丕静,刘晶晶,赵永敏,苏延森,郑春厚
(1.安徽大学 物质科学与信息技术研究院,合肥 230601;2.安徽大学 计算机科学与技术学院, 合肥 230601;3.安徽大学 人工智能学院,合肥 230601)
疾病的关键基因是指在人体系统中与某种疾病密切相关的一组基因,其在人类生理过程和疾病发生过程中具有不可忽视的调控作用,了解关键基因在疾病中的功能和作用,对研究疾病的调控方式、复杂通路、治疗和预后等具有重要意义。关键基因可用于诊断疾病、判断疾病分期、预测和评价新药或新疗法的有效性等。药物靶向治疗的关键是药物能特异性作用于疾病相关基因位点,故识别与疾病紧密相关的关键基因十分重要。但由于基因数量庞大,仅通过生物实验的方法测定基因功能将会耗费巨大的时间成本和经济成本。因此,基于计算模型识别疾病关键基因的预测算法亟待开发。目前有很多研究致力于发现疾病关键基因,此方面研究有助于探索人类复杂疾病的内部发病机制、研究疾病细胞存活所需的最小基因集和后续对疾病的治疗方式及治疗药物的研究[1]。
研究表明,基因并非独立的发挥生物作用,基因之间的相互作用普遍存在,并通过相互作用共同维持着生物内部整体环境的稳定性[2],故基于基因相互作用网络的基因排序技术得到了广泛的应用。Wang等提出了在蛋白质-蛋白质相互作用网络上基于边缘聚类的关键蛋白识别方法,该方法认为节点的重要性由节点与相邻节点之间的相互作用的边缘系数与聚类系数之和决定[3]。Fan等人提出了关键蛋白质预测方法,该方法将亚细胞室信息与基因表达信息相结合,并运用修改后的PageRank算法获得加权蛋白质-蛋白质相互作用网络,实验结果表明其有更好的关键蛋白质预测性能[4]。由此可以看出,将网络拓扑信息和生物学信息结合为研究关键基因提供了很好的思路。然而,虽然目前有多种技术可以用来识别疾病基因,但是大部分方法往往都是通过整合多个样本构建基因共表达网络,弱化了疾病样本与正常样本之间的差异信息,忽略了疾病样本的个体特异性。此外,个体特异性网络构建思想在揭示疾病的个体特征方面已经得到有效的验证[5-6]。
本文以基因间表达相似性为基础构建基因网络,并用来筛选有价值的生物标志物或关键基因,探索基因和疾病之间复杂关系。具体来说,首先利用正常样本的基因表达数据构建参考基因共表达网络,然后依次将每个疾病样本的基因表达数据与正常样本组合,构建疾病样本扰动网络,根据此扰动网络和参考网络,得到每一个疾病样本的个体特异性网络。然后将个体特异性网络作为单层网络,并将单层网络之间的基因联系起来,从而得到多层基因网络,这样既保留了疾病样本的特异性又将多个疾病样本联系在一起。最后,利用Wu等[7]提出的基于张量的多层网络中心性的计算方法,对多层网络中的基因节点中心性进行打分,从而得到关键基因集。与其他经典算法的对比分析表明该方法在预测药物靶标基因上具有一定的优势,功能和通路富集分析证明关键基因集与疾病联系紧密。
1 数据与方法
1.1 数据集
基因表达数据集来源于基因表达综合数据库GEO(https://www.ncbi.nlm.nih.gov/geo/)。本文主要考虑样本量偏少的数据集,因此从GEO数据库中获取哮喘疾病的基因表达数据集GSE31773和GSE43696。在哮喘疾病样本选取的过程中,由于mRNA在CD8+T细胞中的表达差异性大于在CD4+T细胞中,因此选择的疾病样本为CD8+类型的。此外,根据控制变量的原则,尽量使得正常样本和异常样本的其他生物信息如年龄,性别等保持一致。因此,在GSE31773中选取了8个正常样本和6个疾病样本,每个样本包含8 789个基因。同理,在GSE43696中选取20个正常样本和6个疾病样本,每个样本包含9 194个基因。
疾病相关的基因来源于DisGeNet(https://www.disgenet.org/)和Phenopedia(https://phgkb.cdc.gov/PHGKB/startPagePhenoPedia.action)数据库。从两个数据库中获取与哮喘相关的2 712个基因,并与GSE31773和GSE43696数据集中的数据进行整合,分别得到2 522个基因和2 478个基因的表达数据。
此外,从TTD(http://db.idrblab.net/ttd/)数据库获取11个针对哮喘已获批准的药物靶标。
1.2 方法
1.2.1 多层基因网络构建
多层基因网络构建主要分为四步,具体构建过程如图1所示。
第一步是获取疾病相关基因的表达数据。首先从GEO数据库获取正常样本和疾病样本的基因表达数据,从疾病基因相关数据库获取所有与所要研究的疾病潜在相关的基因,从正常样本和疾病样本的表达数据中筛选出疾病相关基因的表达数据。
第二步是利用所有正常样本构建参考基因网络[5]。设参考网络为Gref(V,E,W),其中点集V是由与疾病相关的基因所构成,边集E表示基因对之间的边集,W表示边权,即基因对间的皮尔逊相关系数,其计算方式如式(1)。
ω(ij)=

(1)
其中,Cik表示基因i在第k个正常样本中的表达值,n为基因节点的总数。
第三步是针对每个疾病样本构建个体特异性网络[5]。个体特异性网络的构建参考Liu等[5]提出的方法。具体而言,首先在所有正常样本的表达数据中加入一个疾病样本的表达数据,根据第二步的公式(1),求新的表达数据中基因之间的皮尔逊相关系数,构建一个新的基因网络,并将其看作是加入该疾病样本后的扰动网络[5]。然后根据参考网络和扰动网络构建个体特异性网络,其中边权值为扰动网络和参考网络的边权值的差值绝对值[5]。接着利用拐点分析法设置阈值对网络中的边进行选择,删除一些不显著的边。这种方法考虑到了每个样本的个体特异性,体现了参考网络受到疾病样本的干扰程度,有效衡量了基因间相互作用关系与疾病的相关程度。

图1 多层基因网络构建示意图Fig.1 Schematic diagram of multilayer network construction
注:多层基因网络构建分成四步:第一步是数据收集,即在GEO数据库中选择正常样本和疾病样本,在疾病基因数据库中选择出与哮喘相关的基因;第二步是利用正常样本构建参考网络;第三步是构建疾病样本的个体特异性网络,首先利用疾病样本构建扰动网络,再用扰动网络减去参考网构建个体特异性网络,然后根据阈值去除部分异常边权值后得到最终的个体特异性网络。第四步是整合所有个体特异性网络得到多层基因相互作用网络,层间边连接每层网络中的相同基因,边的权值为1.
第四步是整合单层基因网络得到多层基因网络。将得到的个体特异性网络作为多层基因网络的每一层,依次连接每两个单层网络中的相同节点构建层与层之间的边,边的权值为1,得到多层复用基因网络。
1.2.2 基因节点中心性计算

(2)
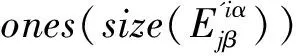
根据单层网络中PageRank算法的幂法求解过程,交互张量H相当于转移概率矩阵,求解张量方程HΦ=λΦ得到中心性二阶张量Φ,其中Φiα表示当前迭代中第α层的第i个基因节点的中心性值,λ表示特征系数,λ这里取值为1,保证二阶张量Φ的存在性和唯一性。迭代结束后,将每个基因节点在所有层中的中心性均值作为该基因的最终中心性值,降序排序后选取排名靠前的基因作为关键基因,分值越高说明基因在疾病中发挥的作用越重要。
2 结果与讨论
2.1 网络边阈值的选取
通过设置皮尔逊相关系数的阈值得到多层网络。具体而言,针对GSE31773和GSE43696两个数据集,分别利用拐点分析法选择拐点,并将其作为筛选边的阈值。根据图2可以发现,GSE31773数据集拐点示意图中,当边权值大于1时,趋势不再有明显上升,因此构建网络的阈值选择为1。同理对于数据集GSE43696阈值选择为0.6。确定数据集GSE31773和GSE43696构建6层网络的阈值分别为1和0.6。


图2 拐点分析图Fig.2 Analysis diagram of inflection point
2.2 多层基因网络有效性分析
利用本文的方法,针对两个独立数据集GSE31773和GSE43696分别构建多层网络,其信息如表1所示,其中层间边连接每层的相同基因,例如数据集GSE43696,其中层间的边数是每层节点连接其他五层中相同节点,即总计37 170条边。以数据集GSE43696构建的多层网络为例,将其可视化后如图3所示。

表1 多层网络信息Table 1 Information of multilayer network
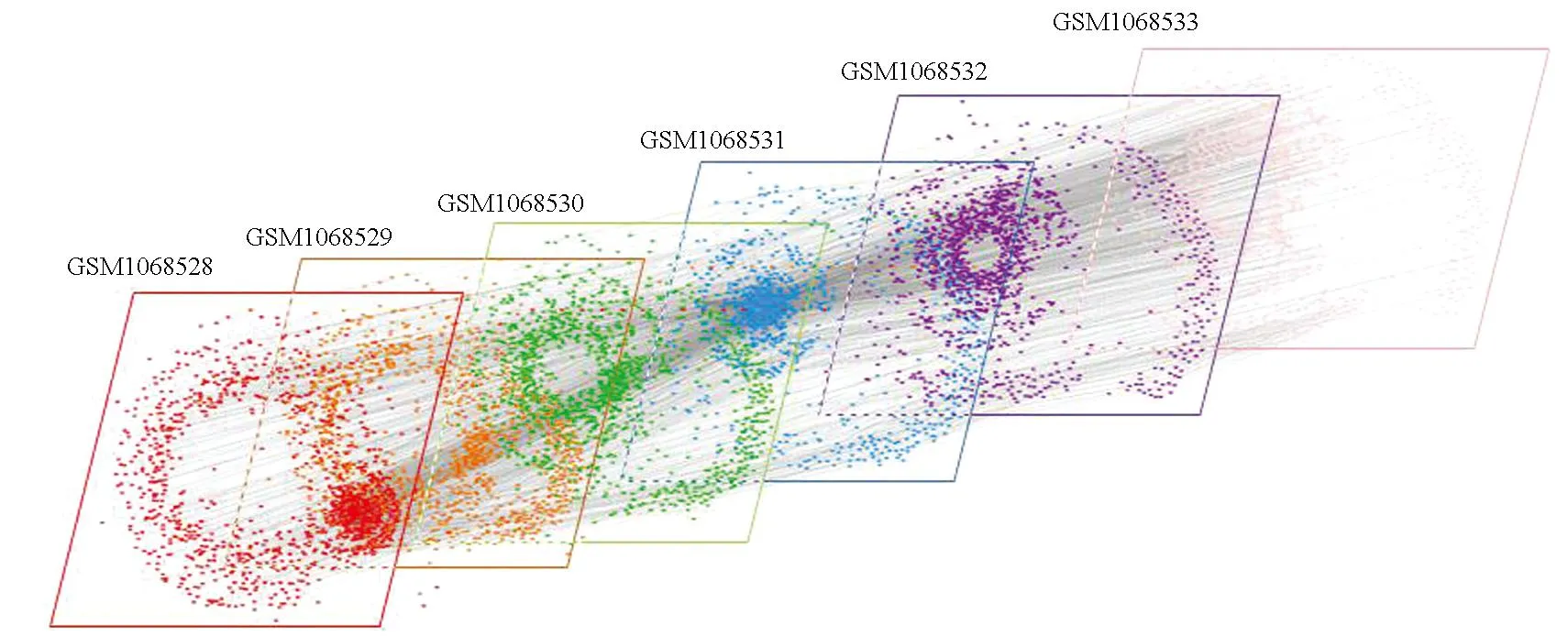
图3 GSE43696: 6层基因网络示意图Fig.3 GSE43696: Diagram of 6-layers gene network
在多层网络构建过程中,多层基因网络的层数有多种选择。为了验证本文构建6层网络的有效性,本文在数据集GSE31773中随机选择不同数量的疾病样本构建了不同层数的多层基因网络,并对比已知哮喘药物靶标基因在不同层数的网络中的排名结果,如表2所示。其中在选择哮喘药物靶标基因时,首先选择有效治疗哮喘的药物,并在数据库中寻找药物关键基因靶标,最终选择包含在本文数据集中的11个靶标基因。表2中“排名1”和“排名2”指随机选择了两次相同数量样本的结果。从表中看出,在六层基因网络中,有5-LOX、IL17、CCR4、IL5RA、ROS等5个哮喘的药物靶标基因排名更靠前;在五层基因网络中,有H1R、IL5、JAK-1等3个基因排名更优,在四层基因网络中,只有基因CAMP有更好的排名;在三层基因网络中,有2个基因JAK-2、IL4R排名更优。综上所述,在识别关键基因集时,构建六层网络的效果更好。
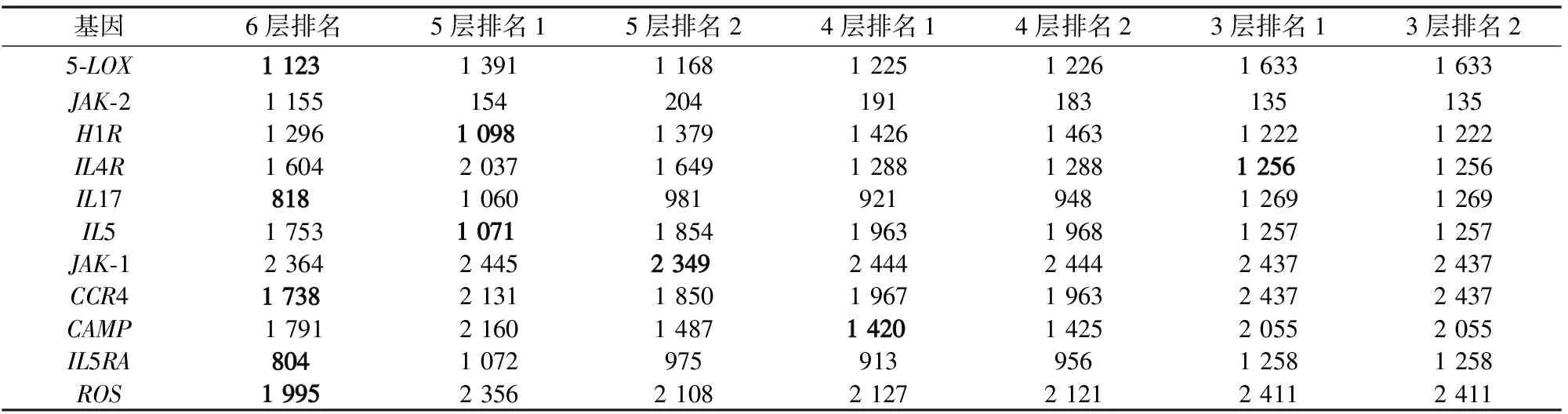
表2 哮喘靶标在不同层网络排名情况Table 2 Ranking of asthma targets in different layers of networks
2.3 算法对比分析
根据哮喘基因数据集,利用本文提出的方法,可以得到哮喘相关的基因。为了进一步评估已知的疾病特异性通路或基因是否在预测的关键基因上具有显著的优先级,本研究利用GSEA软件的GSEAPreranked工具对结果进行分析。GSEA富集分析主要是用来评估一个预先定义的基因集在与表型相关的基因排序列表中的分布趋势,它不需要进行基因过滤,输入数据主要包括两部分,一种是预先定义的基因集,一种是给定的基因排序列表。本文中,预先定义的基因集是KEGG通路数据库中的哮喘特异性相关的基因集,基因排序列表是本文预测的所有基因排序结果。通过GSEA富集分析揭示我们的模型结果和KEGG通路数据库中哮喘特异性相关的基因集之间的关联,以GSE31773数据集为例,根据其所有基因排名和KEGG通路数据库中哮喘特异性相关的基因集进行加权K-S检验得到p值,结果如图4所示。结果表明,与其他预测关键基因的排序方法MI[8],t-Test[9],PCC[10],SCC,FC[11],NetRank[12],MarkRank[13]相比,本研究中的算法在对疾病关键特异性基因进行优先排序时具有显著的p值。
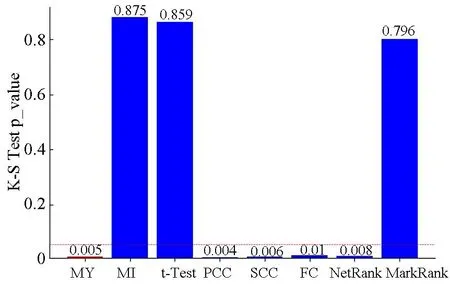
图4 不同对比算法的哮喘通路富集分析Fig.4 Enrichment analysis of asthma pathways withdifferent comparison algorithms
2.4 哮喘关键基因集分析
为了验证本方法所识别的疾病相关基因的重要性,针对GSE31773和GSE43696两个数据集,分别选择排名前10的关键基因(见表3),分析是否已有研究证实其为哮喘关键基因。研究发现TP53、MAP3K1、COL18A1、DACT1、CD40LG、ANKRD55、CD4以及TNFSF18、AFM、NKX2-1、SCGB1A1、RAG1、FRAS1、HSD11B2、GSTO2、SOAT1、IL19等基因在哮喘发生发展过程中起重要作用。例如,Yuan等[14]的研究表明,与迟发性哮喘临床表型相关的TP53差异甲基化位点是早期筛选的有效生物标志物。Zhang等的研究证明DACT1可能是治疗哮喘的潜在靶点[15]。对于CD40LG,有研究表明CD86和CD40LG之间的相互作用会促进过敏性哮喘的发展[16]。CD4T细胞淋巴细胞活化在严重哮喘发病机制中起重要作用[17]。SCGB1A1是肺重要的防御分子,防止SCGB1A1被抑制可有效的改善哮喘[18]。有研究表明GSTO2是哮喘易感基因,GSTO2基因的多态性和哮喘有关[19]。此外,有研究证实,IL-19基因在哮喘中高度表达,在变应性疾病中起着重要作用[20]。研究还发现,嗜酸性粒细胞的凋亡在支气管哮喘病理生理中发挥至关重要的作用[21],并且PUS10基因能够调节Trail诱导的细胞凋亡过程[22]。轴突或突触结构调控哮喘的激发机制[23],且F5蛋白在膜-细胞骨架相互作用和突触结构或功能的动态方面发挥重要作用[24]。此外,“下丘脑-垂体-肾上腺”轴功能与肺功能改善程度相关[25],并且CRHBP调节促肾上腺皮质激素控制“下丘脑-垂体-肾上腺”轴功能[26]。由此推断,PUS10、F5及CRHBP等基因也与哮喘发生发展紧密相关。
2.5 GO功能富集分析
为了分析本算法预测的疾病关键基因的功能相关性,利用本算法分别在两个独立数据集上选择排名前100的基因,使用基因功能分析工具DAVID对其作GO功能富集分析。基于DAVID分析工具,得到与前100个基因显著相关的基因本体,图5展示了排名前10的基因本体。图的纵坐标展示了GO的功能注释,横坐标上的值表示GO在关键基因集中的富集显著性值-log(p)。由图可以发现,在排名前10的基因本体中,免疫反应、调控T细胞增殖、T细胞刺激以及细胞因子活性均被证实与哮喘有密切联系[38]。具体来说,哮喘是由免疫系统对环境因子和不同的基因表达的联合反应引起的呼吸系统疾病。T细胞是哮喘中过敏性气道炎症的关键介质[39],T细胞的增殖会引起免疫球蛋白水平增加和支气管高反应性即哮喘发作,细胞因子也会辅助T细胞增殖的反馈控制。此外,炎症反应也与哮喘相关,在哮喘恶化过程中伴随着循环嗜酸性粒细胞、嗜碱性粒细胞及其前体细胞的变化等各种炎症反应[40-41]。除上述机制外还有几种潜在的新机制,例如药物反应,内皮细胞分化,蛋白质磷酸化调控,信号调控,应对缺氧,转录调控等在哮喘发展过程中都起着重要的作用。
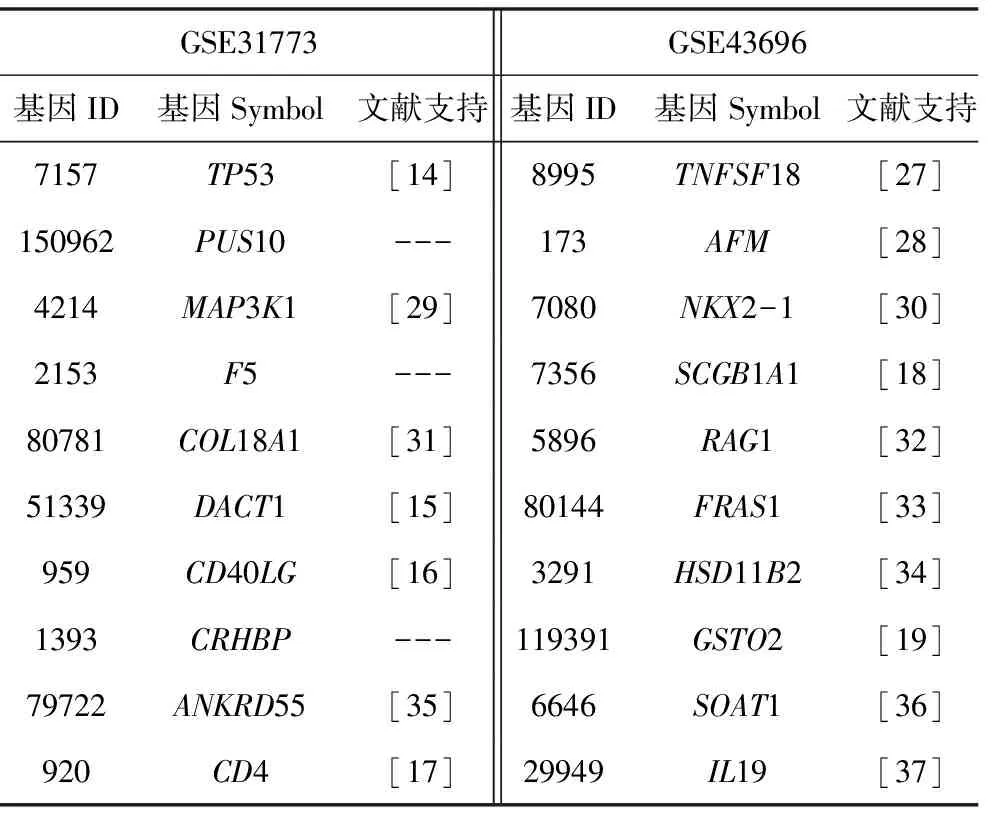
表3 排名前10的关键基因集Table 3 Top 10 critical gene sets
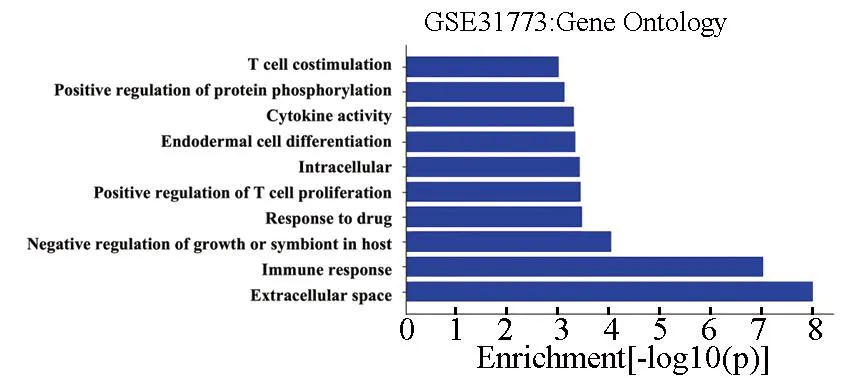

图5 关键基因富集Gene OntologyFig.5 Key gene enrichment Gene Qntology
2.6 通路富集分析
为了定位关键通路的关键基因,本文基于DAVID平台对两个独立数据集排名前100的基因进行通路富集分析,得到与100个基因显著相关(p_val≤0.05)的通路,表4和表5展示了显著相关的通路。由上述通路富集分析结果可知,细胞因子受体相互作用、趋化因子信号通路、T细胞受体信号通路、原发性免疫不全四条通路都与哮喘紧密相关。肿瘤坏死因子(TNF)信号通路、TGF-beta信号通路、Th1/Th2分化等通路也被证明与哮喘有关。TNF信号通路的坏死因子α是免疫和炎症反应的有效调节剂,可以引起包括哮喘在内的多种自身免疫性疾病[42]。哮喘会通过TGF-beta信号通路促进小鼠脉络膜血管新生[43]。T淋巴细胞介导的对过敏原的免疫应答是哮喘发病机制的早期关键因素,而Th1/Th2平衡是哮喘发病机制的核心[44]。此外,还有若干个与哮喘潜在相关的通路,包括黏着连接、焦点粘连、鞘脂类信号通路等。

表4 GSE31773:关键基因通路富集分析Table 4 GSE31773: Pathways enrichment analysisof critical genes
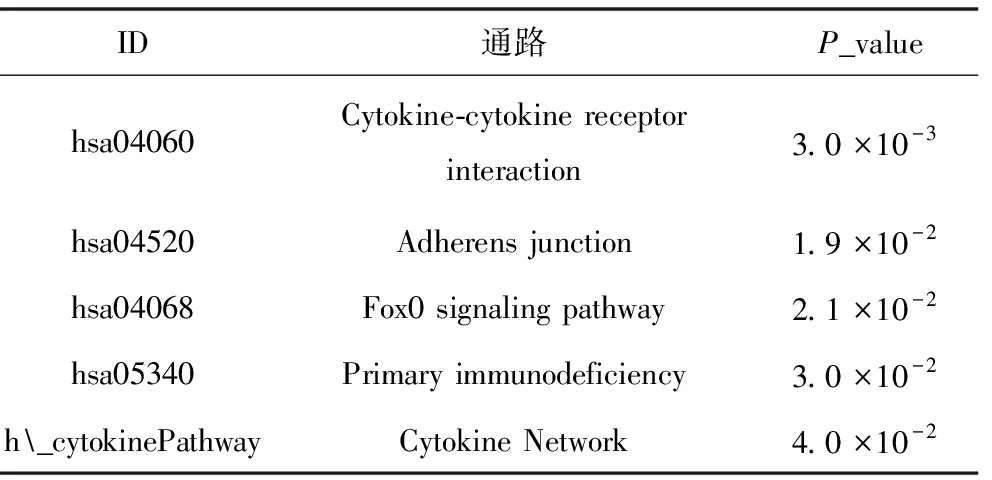
表5 GSE43696:关键基因通路富集分析Table 5 GSE43696: Pathways enrichmentanalysis of critical genes
3 结 论
1)复杂疾病的发生发展本质上与基因和生物功能过程的改变密切相关,疾病关键基因的识别对于研究疾病机理尤其是药物靶向治疗具有重要意义。哮喘作为全球范围内发病率最高的慢性呼吸道疾病之一,其发病率在逐年上升。识别出与哮喘成因紧密相关的基因有助于提高治疗效果。然而临床研究中由于疾病样本数较少,通常导致疾病相关基因识别困难。针对上述问题,本研究提出基于少数样本构建多层网络,进而利用多层网络随机游走识别疾病相关的关键基因的方法。该方法有助于挖掘样本数量受限条件下的疾病相关基因,加深对疾病致病机理的理解。
2)构建的多层网络对识别小样本疾病的致病基因可行且有效。本文利用皮尔逊相关系数计算出每条边的权值;为增强网络结构的稳定性,采用拐点分析法寻找最佳阈值,保留扰动程度较大的边;通过比较对已知疾病关键基因的排序选取最优的网络层数。例如针对数据集GSE31773的实验分析表明,构建六层基因网络效果最佳。
3)与其他方法相比,本算法识别的哮喘相关基因的排名更具显著性。利用本算法分别在GSE31773和GSE43696数据集中挖掘排名前10的关键基因,研究发现TP53、MAP3K1、COL18A1、DACT1、CD40LG、ANKRD55、CD4以及TNFSF18、AFM、NKX2-1、SCGB1A1、RAG1、FRAS1、HSD11B2、GSTO2、SOAT1、IL19等基因在哮喘发生发展过程中起重要作用,并推断PUS10、F5及CRHBP等基因也与哮喘发生发展紧密相关。
4)对分别从GSE31773和GSE43696两个数据集中所得排名前100的关键基因进行通路富集分析和GO功能富集分析,分析结果表明所识别的基因能够显著富集到与哮喘相关的通路和功能中。
