α2珠蛋白基因Questembert变异联合-α3.7 缺失导致α-地中海贫血一家系的遗传学研究
2024-01-11张鑫丽阳鑫妙唐克锋李雯雯沈国松
张鑫丽 阳鑫妙 唐克锋 李雯雯 沈国松
α-地中海贫血是由于α-珠蛋白基因缺陷导致的α-珠蛋白链合成障碍引起的遗传性溶血性贫血,它是最常见的常染色体单基因遗传病,其基因缺陷包括大片段丢失(占95%)和α-珠蛋白基因血红蛋白A1 带(HbA1)、血红蛋白A2 带(HbA2)变异。病情严重程度与α-珠蛋白链减少的程度直接相关[1]。本研究对1 例严重溶血性贫血疑似α-地中海贫血的患儿和家系进行致病基因分析,发现了中国人群罕见的α-地中海贫血致病型变异,并对先证者母亲羊水进行产前诊断,现将结果报道如下。
1 对象和方法
1.1 对象 先证者,男,4 岁,贫血严重。2021 年5 月20 日因母亲再次妊娠至湖州市妇幼保健院遗传咨询门诊就诊。患儿为足月分娩,母孕期和患儿出生时无异常,患儿出生后1 个月,呈现贫血状态,2 个月起开始输血治疗,现每4 周输血400 mL,42 个月时行脾切除手术。患儿中度贫血貌,皮肤黏膜黄染出血。父母非近亲婚配,父亲籍贯江苏,母亲籍贯贵州,父亲表型正常;母亲此次为自然妊娠中期,胎儿B 超检查显示为双绒双羊双胎,无其他异常;母亲及其家系有多名贫血及脾肿大或脾切除史者。收集患儿父母及母亲家系贫血成员相关病历,并进行血液学相关检查。本研究经本院医学伦理委员会审查通过(批准文号:2022-J-110),患儿父母与家系成员均签署知情同意书。
1.2 方法
1.2.1 血常规和Hb 电泳检测 收集患儿、患儿父母及母亲家系部分成员非空腹外周静脉血2 mL,EDTA 抗凝。先采用全自动血液分析仪进行血常规测定,再采用Sebia capalliary2 毛细管电泳仪进行Hb 成分分析。
1.2.2 常见地中海贫血基因检测 抽取患儿和父母的非空腹外周静脉血2 mL,EDTA 抗凝。母亲孕18 周穿刺取羊水10 mL,进行DNA 提取及荧光定量PCR 实验。试剂由厦门致善生物技术有限公司提供,采用“多色荧光PCR 熔解曲线(MMCA)”技术,按照基因诊断试剂盒的操作说明在罗氏荧光定量PCR Lightcycler 480Ⅱ上进行中国人常见α-地中海贫血基因(包括3种缺失型变异:-SEA、-α3.7、-α4.2,3 种常见非缺失型变异:HbCS、HbWS、HbQS)检测。
1.2.3 血液系统疾病Panel 检测 将患儿、患儿父母及母亲家系成员的外周血及母亲羊水送至北京迈基诺医学检验所进行检测。根据该所提供的血液系统疾病Panel 检测项目,从先证者样本中提取DNA,构建文库,使用IDTxGen®Exome Research panel v1.0 杂交捕获试剂完成目标区域捕获,采用Illumina NovaSeq 完成高通量测序(测序读长为2×150 bp)。通过生物信息学分析,并结合当前国内外现有的行业指南和医学知识,筛选出与表型相关的靶向外显子组区域的(可能)致病性变异,并对其设计引物,进行Sanger 测序验证,引物序列HbA2-F:5'-CAGGATTGGGCGAAGCCCT-3',HbA2-R:5'-GAGGTCCTTGGTCTGAGACAGG-3';并对父母、母亲家系及母亲羊水进行该变异位点的Sanger 验证。实验过程中,对检测样本的质控分析贯穿整个过程,并选用8~10 个高频单核苷酸多态性(single nucleotide polymorphisms,SNP)位点作为样本身份标识。
2 结果
2.1 先证者及家系成员检测结果 见表1。
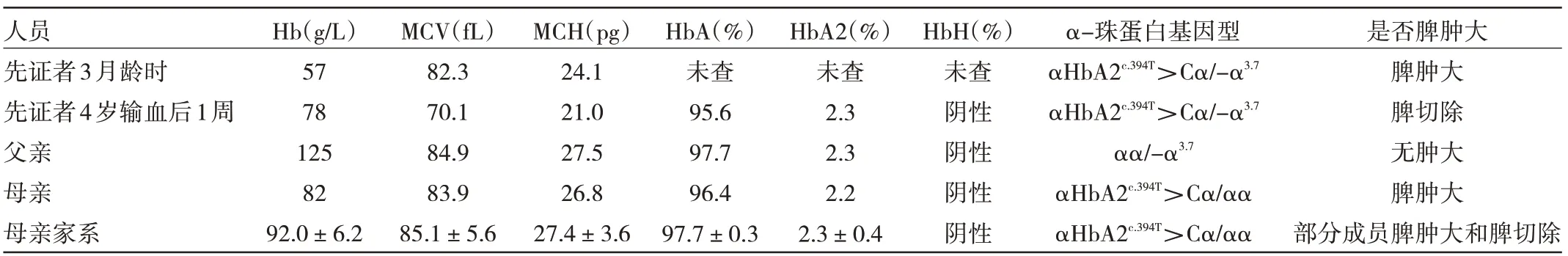
表1 先证者及家系成员检测结果
2.2 常规地中海贫血基因诊断 结果发现,先证者和其父亲存在αα/-α3.7杂合缺失型突变。
2.3 DNA 变异信息 二代测序后提示先证者HbA2 基因3 号外显子处有一个纯合突变HbA2c.394T>C(p.S132P),Sanger 测序验证母亲携带该位点的杂合性变异,父亲为野生纯合,见图1。母亲家系的部分成员进行该位点验证,发现了8 名携带者;母亲羊水细胞Sanger 测序双胎均未携带此变异位点,见图2,随访为健康双胎女婴。
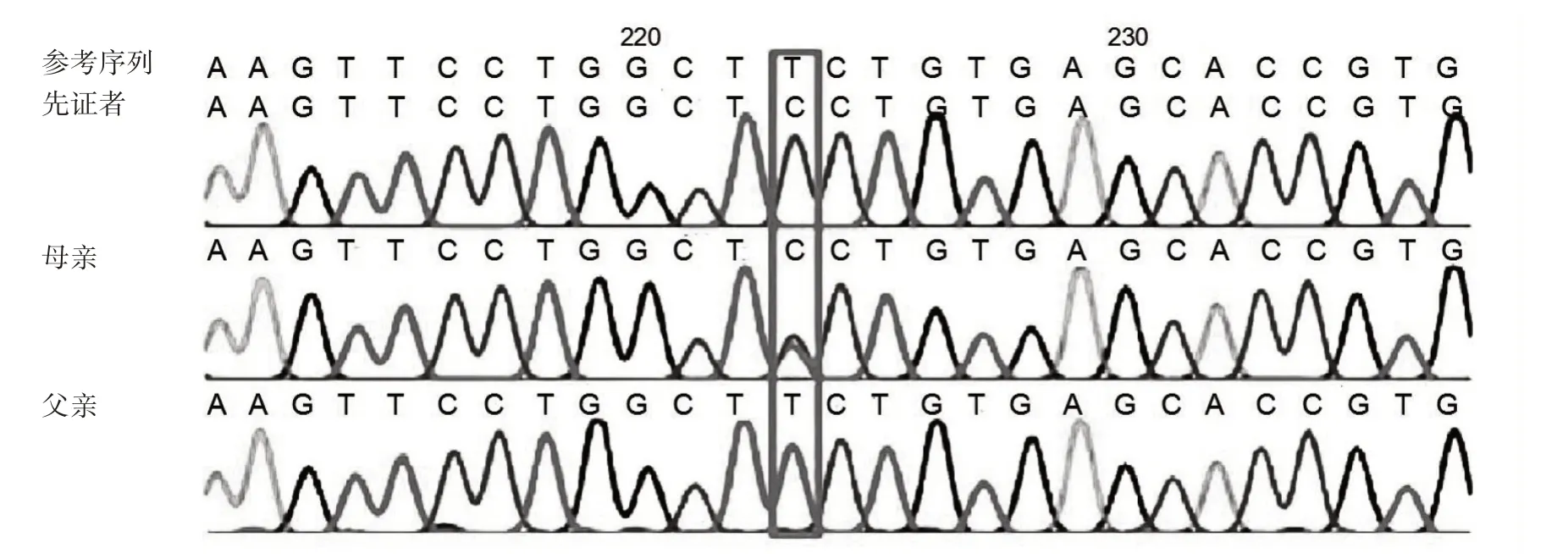
图1 HbA2 基因c.394T>C 变异的Sanger 测序图
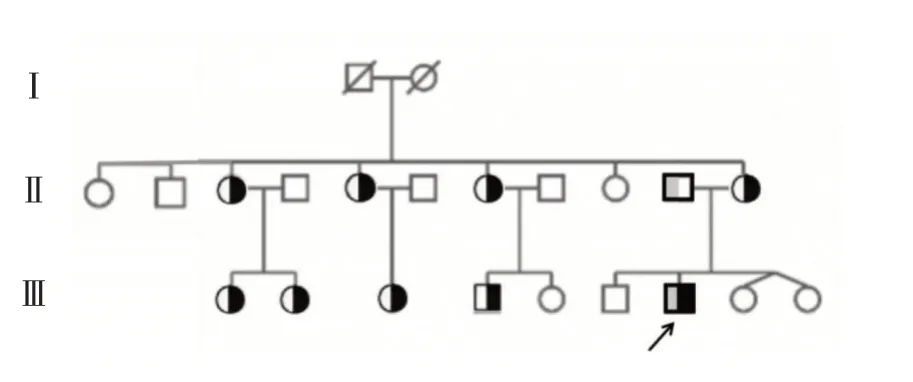
图2 患儿家系系谱图
3 讨论
α-珠蛋白基因位于16 号染色体上(16p13),每条染色体有2 个连锁的α-珠蛋白基因,即HbA1 和HbA2基因。1 个或2 个α-珠蛋白基因的合成缺陷会导致红细胞参数发生轻度到中度的变化。大多数已确定的α-地中海贫血患者是由1 个或2 个重复α-珠蛋白基因的大量缺失引起,这是主要的致病突变,其余则是HbA1 和HbA2 基因点突变引起的非缺失型地中海贫血。此外,已经报道了超过100 种不同的点突变。这些点突变如寡核苷酸插入和缺失大多与α-珠蛋白基因正常表达的关键基因组区域相关[2],通过多种机制减少或抑制α-珠蛋白链的合成,如影响转录、剪接、翻译、翻译后加工等[3]。而一些由α-珠蛋白基因等位基因共同遗传的非缺失型变异造成α-珠蛋白不稳定变体的患者可能出现与地中海贫血中间型血红蛋白H带(HbH)病[4]相似的临床和血液学结果。其中产生极不稳定的α-珠蛋白变异体的,临床症状(贫血、肝脾肿大、输血频次)更加严重[5]。因为不稳定的α-珠蛋白变异体可能没有Hb 运输释放氧气和二氧化碳的作用,但它们具有高氧亲和力,导致组织供氧减少,造成溶血和贫血,最终导致造血不良的骨髓扩张,骨、肝、脾的髓外造血[3]。
第三外显子突变引起第三外显子编码区域的螺旋H 的破坏,最终造成不稳定的α2 珠蛋白链变体比较多见。H 螺旋从118 个氨基酸残基延伸到138 个氨基酸残基,在血红蛋白四聚体中α1 和β1 单体的疏水接触中起关键作用,或在α-珠蛋白稳定蛋白(AHSP)(α-珠蛋白的分子伴侣)的形成中起关键作用[6]。H 螺旋的突变产生极不稳定的Hb 变体[1],并在红细胞中沉淀,导致溶血,出现α 地中海表型[7-8]。如Hb Toyama(p.Leu135Arg)已证实其表达水平低于总Hb 的1%[9];Hb Quong Sze(p.Leu125Pro)是在1 例HbH 病患者中发现的[10];Hb Sun Prairie(p.Ala130Pro)[11]和Hb Attleboro(p.Ser138Pro)[12]为两种低水平表达的不稳定Hb 导致患者出现严重的溶血。
而HbA2 基因c.394T>C(p.Ser132Pro),又称为Hb Questembert 变异,可引起H 螺旋中132 位极性中性氨基酸(丝氨酸)转变为非极性疏水性氨基酸(脯氨酸),导致H 螺旋结构和稳定性的破坏。迄今为止该变异有少量报道,其表型主要是结合了地中海贫血和周围溶血性贫血的特征。该变异首次于1993 年在法国Questembert 地区的一个先天性Heinz 小体贫血8 例患者的家系中被报道[13]。该家系为HbA2 基因c.394T>C变异,功能试验证实该变异影响了从mRNA 转录、剪接到蛋白的空间结构及转录和翻译过程,产生了极不稳定α-珠蛋白链,且其在红细胞中表达水平非常低,造成患者造血功能障碍和周围性溶血。另1 例报道为希腊雅典的2 岁重度溶血性贫血患儿,其HbA1 基因的c.394T>C 变异,并且在反式位置检出-α3.7缺失[14]。
本研究中,Hb Questembert 变异在家系中携带者表现为轻到中度地中海贫血,地中海贫血特征要严重于αα/-α3.7缺失型携带者。而先证者测序为HbA2 的c.394T>C 的纯合变异是由于合并反式-α3.7缺失造成,其α-地中海贫血基因型为αQuestembertα/-α3.7。由于HbA2基因编码的mRNA 的稳定水平比HbA1 基因高3 倍[15],HbA2 上的点突变对α-珠蛋白链影响可能更大[13]。本例先证者对比希腊雅典患者,具有典型的地中海贫血血液学特征和严重的临床表现,其表型倾向于非缺失型HbH 病。
地中海贫血非缺失型变异携带者Hb 成分很多没有明显的变化,常规的PCR 法也难以检测。然而,随着分子诊断技术的发展,许多α-地中海贫血病的鉴定变得更加容易。因此,临床上要对地中海贫血保持高度敏感性,必要时应用遗传咨询,以降低母亲怀孕期间严重的并发症和杜绝中重型α-地中海贫血患儿的出生。Hb Questembert 变异首次在我国被发现,丰富了中国人群α-地中海贫血突变谱,且经过家系验证,该变异可导致典型的α-地中海贫血,为该家系在遗传咨询和产前诊断中提供重要依据。
遗传性血红蛋白病,包括α-地中海贫血症的遗传咨询是至关重要的,至少可以最大限度地减少巴特血红蛋白胎儿积液综合征的发生,该综合征可能导致新生儿死亡和怀孕期间母亲的严重健康并发症。只有当临床结果准确区分α-珠蛋白基因型和其他相关基因型,且表现与各种形式的α-地中海贫血相同时,遗传咨询才有意义。随着遗传咨询技术的发展,许多α-地中海贫血病遗传修饰因子的鉴定变得更加容易,并记录了它们对各种表型的影响。
