冬季农田不同深度土层及小麦根际土壤细菌群落结构和共现网络分析
2024-01-10田泽远张俊霞张文俊顾效纲胡红伟
田泽远,张俊霞,张文俊,刘 彪,顾效纲,胡红伟
(1.河南城建学院河南省水体污染防治与修复重点实验室,河南 平顶山 467036;2.江西理工大学资源与环境工程学院,江西 赣州 341000)
土壤细菌多样性对维持生态系统功能至关重要。土壤细菌群落具有驱动地球化学循环、分解有机物、抑制土传疾病和促进植物生长等功能[1]。影响土壤细菌群落结构的因素有很多,如pH、有机质、氨和磷等[2]。土壤细菌群落的组成与土壤深度也相关。在土壤深处,细菌可利用的养分有限,生物量随着深度增加呈指数下降[3],但某些细菌的潜在活性可能高于表层土壤[4]。此外,即使在相隔几微米至几毫米的土壤环境中,物理和化学性质、细菌丰度、群落组成和细菌活性也可能存在较大差异[5]。
植物根际是土壤细菌进行物质转化、营养吸收的重要场所。根际细菌有助于植物在养分同化之前将其转化为可用的形式[6]。同时,植物根系能够产生如氨基酸、有机酸、糖类和次生代谢产物等物质,被土壤细菌用于供能和生物量生产,从而促使更多有益细菌在土壤中定殖,形成密集复杂的根际微生态[7]。植物-土壤反馈是细菌群落结构的重要决定因素,尤其在农业土壤生态系统中。Fan等[8]通过对小麦根际细菌的研究发现,非根际土壤和根际土壤的细菌群落组成差异显著,并且细菌多样性与根系的距离成反比。此外,与非根际土壤相比,根际细菌共现网络结构相对简单[9]。
冬季是小麦休眠期,小麦停止生长后,将营养成分储存在根部,为春季返青提供足够养分,这一过程对小麦产量至关重要。研究这一时期农田土壤及小麦根际土壤微生物种群结构及共现网络关系,将有助于深入了解冬季小麦根际微生态及其响应机制。本研究选择河南省平顶山市汴城村附近农田土壤及小麦根际土壤为研究对象,使用Illumina Miseq平台进行高通量测序,选择16SrRNA V4区进行PCR扩增,对细菌群落的多样性及组装机制进行了探究。
1 材料与方法
1.1 采样区域概况与样品采集
采样点位于河南省平顶山市汴城村附近农田(33°40′53.05"N,113°17′15.17"E),该区域属大陆性季风气候,年降水量500~600 mm。农田为多年小麦-玉米连作耕地,土壤类型为褐土,质地为壤质土。2020年1月,采用五点取样法,使用直径为5.5 cm的空心杆手动螺旋钻采集垂直深度为0~20 cm的农田土壤样品,标记为非根际(FG)样品(0≤FG-A≤5 cm、5<FG-B≤10 cm、10<FG-C≤15 cm、15<FG-D≤20 cm),在各点附近采集小麦根际土壤,标记为GJ样品(GJ:4~10 cm)。将同类型土壤样品混合均匀后,分为两份,样品保存于-40℃超低温冰箱。
1.2 土壤样品理化性质测定
土壤样品自然风干并过100目筛网后用于理化参数测定。参照《土壤农业化学分析方法》[10]测定土壤中的氨氮(NH+4-N)、硝酸盐(NO-3-N)、亚硝酸盐(NO-2-N)、总磷(TP)和总有机碳(TOC)。采用浓H2SO4-H2O2法消解,使用凯氏定氮仪(KjeFlex K360,Switzerland)测定土壤凯氏氮(TKN)。采用电位法,使用pH计(Hanna HI98130,Italy)测量pH值。
1.3 PCR扩增和高通量测序
称量0.1 g土样,采用E.Z.N.ATMMag-Bind Soil DNA Kit试剂盒提取土壤样品DNA,采用1%琼脂糖凝胶电泳检测DNA提取质量,采用紫外分光光度计进行定量测定。利用16S rRNA V4区引物进行PCR扩增,引物序列分别为515F(5′-GTGCCAGCMGCCGCGGTAA-3′)和806R(5′-GGACTACVSGGGTATCTAAT-3′)。使用Illumina Miseq平台进行高通量测序,高通量测序由生工生物工程(上海)股份有限公司完成。本研究所获数据已上传至NCBI,SRA序列号为PRJNA776557。
1.4 数据分析
基于97%相似度,使用Usearch 7.1对高通量测序结果进行OTU(Operational Taxonomic Unit)分类,在聚类过程中去除嵌合体,得到OTU代表序列[11]。使用Mothur 1.30.1进行细菌群落Alpha多样性分析。使用R的vegan包进行细菌群落Beta多样性分析和冗余分析(RDA)。使用SPSS25.0对数据进行皮尔逊线性相关性分析。使用R的hmisc和igraph包分析细菌群落之间的相关性,并使用Gephi 0.9.2(R>0.6,p<0.05)进行网络可视化。利用Excel 2019进行实验数据的统计与计算。
2 结果
2.1 土壤样品理化性质
土壤理化参数如图1所示。土壤pH值介于4.33~5.81,呈酸性,并随土壤深度增加而增加。TOC含量在各样品之间存在显著差异(p<0.05),且随土壤深度的增加而减少,GJ中含量最高。TP含量在各样品之间差异不显著,介于1.40~1.60 g/kg。NO-2-N和NO-3-N含量最高值分别出现在FG-D(22.4 mg/kg)和FG-B(6.91 g/kg),均显著高于其他样品(p<0.05)。GJ样品中NH+4-N和TKN含量均显著高于其他样品(p<0.05),分别为58.4 mg/kg和0.72 g/kg,并随土壤深度增加而减少。
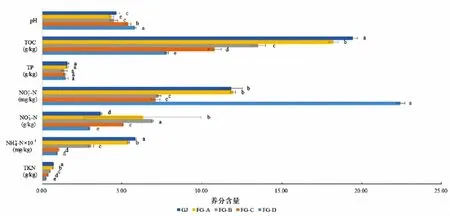
图1 土壤样品理化参数
2.2 土壤样品细菌多样性
土壤样品Alpha多样性指数如表1所示。文库覆盖率均大于94%,说明取样合理,样本OTU覆盖度已经趋近饱和。GJ样品OTU数目最少,最高值出现在FG-C。香农指数最低值和辛普森指数最高值均出现在GJ,表明GJ样品中细菌多样性最低。根据香农指数和辛普森指数变化趋势,可知细菌多样性随着土壤深度增加而增加。此外,基于OTU的样本聚类树分析(见图2),不同样本细菌群落可分为两个大类,即GJ、FG-A和FG-B聚类,FG-C和FG-D聚类。
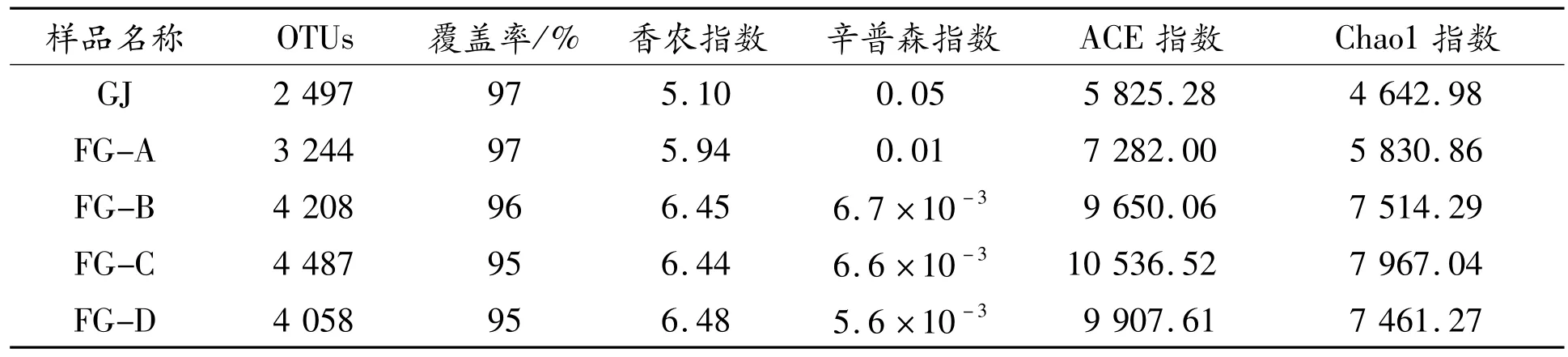
表1 土壤样品Alpha多样性指数

图2 基于OTU的样本聚类图
2.3 土壤样品细菌群落组成
基于门水平的微生物群落相对丰度前10的物种,绘制物种相对丰度柱状图(见图3(a))。变形菌门(Proteobacteria)、酸杆菌门(Acidobacteria)、放线菌门(Actinobacteria)和厚壁菌门(Firmicutes)在所有样品中均为优势菌门。其中,变形菌门在GJ(38.79%)、FG-A(51.03%)和FG-B(40.58%)中丰度最高;酸杆菌门在FG-C(30.58%)和FG-D(31.65%)中丰度最高。相较于农田土壤柱状样,小麦根际土壤样品中放线菌门的丰度明显高于土壤柱状样中放线菌门的丰度,而酸杆菌门则相反。此外,在柱状样中,变形菌门和拟杆菌门的丰度随土壤深度增加而减少,而酸杆菌门则呈相反趋势。
基于属水平的微生物群落相对丰度前20的物种,绘制物种相对丰度柱状图(见图3(b))。Gp6、节杆菌属(Arthrobacter)、Gp4、芽单胞菌属(Gemmatimonas)是所有样品中的优势菌属。其中,节杆菌属在GJ(19.21%)中丰度最高,芽单胞菌属在FG-A(4.59%)中丰度最高,属于酸杆菌门的Gp6在FG-B(5.21%)、FG-C(34.79%)和FG-D(37.53%)中丰度最高。相较于农田土壤柱状样,小麦根际土壤样品中节杆菌属的丰度最高,而Gp6的丰度最低。此外,在柱状样中,Gp6的丰度随土壤深度的增加而增加,而Gp4的丰度则随土壤深度的增加呈现出先增加后降低的趋势。
2.4 土壤细菌群落结构与环境因子的相关性分析
对土壤细菌群落结构与环境因子进行RDA分析(见图4),RDA1解释总变异量的69.26%,RDA2解释总变异量的26.38%,二者共同解释了细菌群落变异量的95.64%,能够较好地反映出微生物群落与环境因子之间的相互关系。由图4可知,样品GJ、FG-C和FG-D细菌群落与pH呈正相关,且FG-C和FG-D聚类。FG-A、FG-B细菌群落与pH呈负相关,与TKN、TOC和NH+4-N呈正相关。
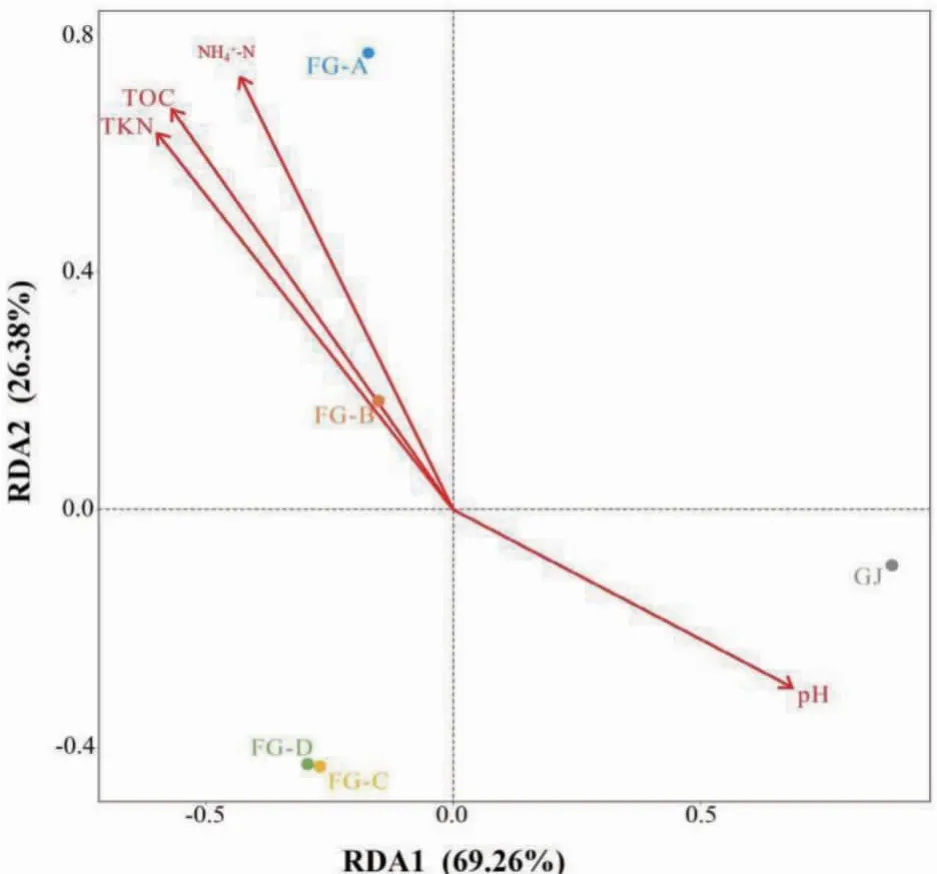
图4 细菌群落与环境因子之间的RDA分析
2.5 细菌群落共现网络分析
使用共现网络分析评估前150 OTUs的细菌相互作用(见图5)。该网络生成了98个节点和1 527条边,平均度为31.16,网络直径为9,模块度为1.27,平均聚类系数为0.76,平均路径长度为3.67(见图5(a))。基于显著的斯皮尔曼相关性(ρ>0.6,p<0.05),总共预测了7个模块(见图5(b))。每个节点的大小与连接程度成正比;边的粗细与斯皮尔曼相关系数成正比。绿色线表示显著负相关,红色线表示显著正相关。节点越大,表明它与其他节点关联越多,连线越宽,表明两节点之间的相关性越强。基于此,与其他10个以上节点有紧密联系的OTU被定义为关键分类群,表明它们在每个细菌群落的组成和功能方面均具有重要的作用(见图5(b))。本研究中,Povalibacter、假单胞菌属(Pseudomonas)、鞘氨醇单胞菌属(Sphingomonas)、Gp6、Gp4、Gaiella、Subtercola等节点是网络主要模块中最重要的基础单元分类群,表明这些细菌为细菌群落中的优势细菌并在其中起关键作用(见图5(a))。
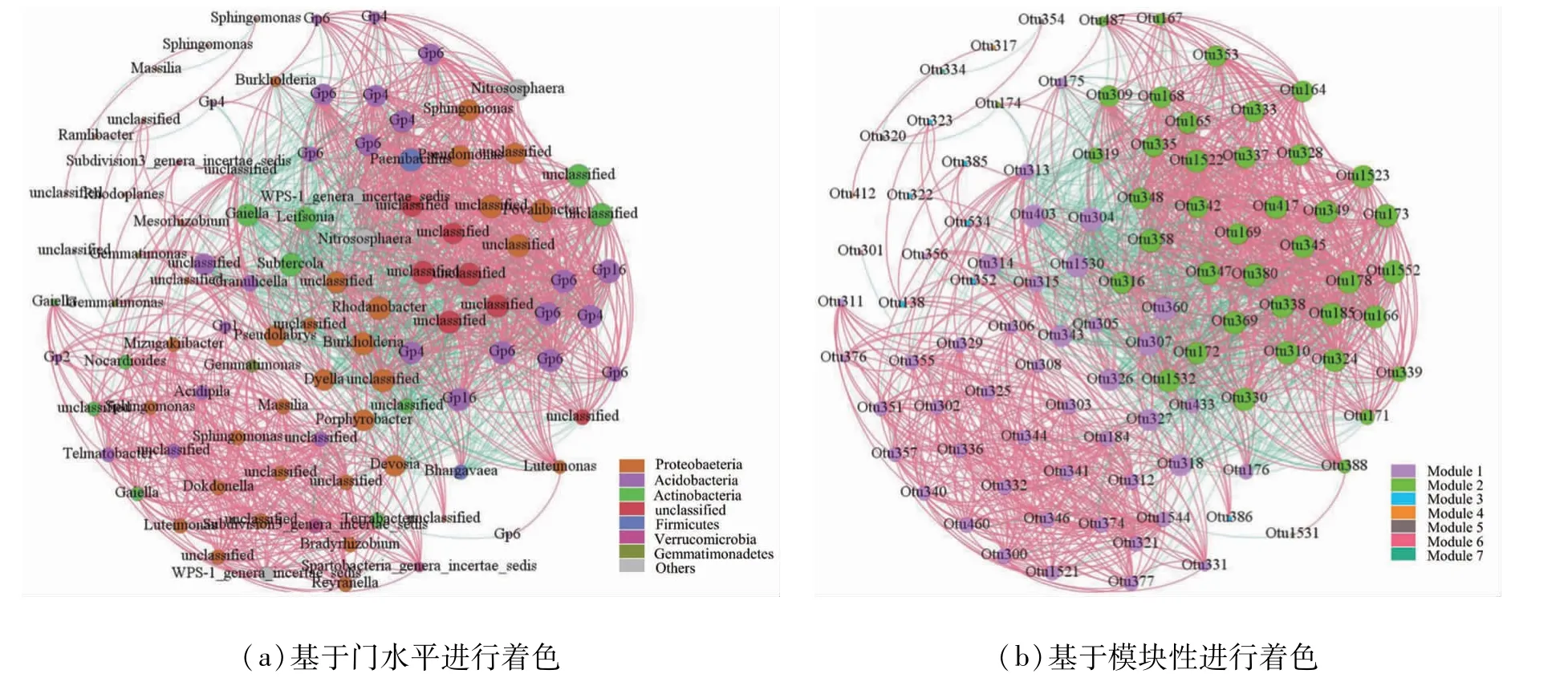
图5 基于全部土壤样本中前150个OTUs的共现网络图
3 讨论
样品采集区域土壤为褐土,与其他类型土壤相比,褐土有机质和全氮含量比较低。对于作物而言,褐土缺乏氮素营养[12],在作物播种前一般需对农田施肥。本研究中5个土壤样品pH值介于4.33~5.81,呈酸性。这主要是因为农田土壤长期施用氮肥,在氮肥转化为硝酸盐的过程中,导致土壤中Ca、Mg等碱性离子流失,从而引起土壤pH降低[13]。理化性质和空间分布能够极大地影响细菌群落的组装,从而导致细菌群落丰富度和多样性的差异[14]。由RDA分析可知,FG-A在TKN、TOC、NH+4-N上的投影距箭头较近,表明FG-A中细菌群落对土壤中TKN、TOC、NH+4-N的响应较为敏感,而GJ在pH上的投影距箭头较近,表明pH对GJ中细菌群落结构影响较大。
结果表明,变形菌门、酸杆菌门、放线菌门和厚壁菌门为优势菌门,这与Li等[15]和Chang等[16]对农田的研究结果一致。不同于非根际土壤样品,在小麦根际土壤中,变形菌门丰度最高,其次为放线菌门,与Fan等[17]的研究结果相似。此外,变形菌门在FG-A和FG-B中丰度最高。一般认为变形菌和放线菌是富营养型细菌,对土壤中C、N循环有显著的促进作用[18]。植物根际土壤细菌群落结构常表现出与非根际土壤之间的显著差异,而本研究中基于OTU的样本聚类结果显示,GJ、FG-A和FG-B之间相互聚类,表明它们的细菌群落结构特征相似,可能是因为小麦在冬季处于休眠期,根系分泌物较少,对根际细菌的影响有限[19],且该时期小麦根系长度约为10 cm,与FG-A和FG-B土壤样品处于同一深度。可见,本研究中土壤深度是冬季农田土壤细菌群落结构的主要影响因子。
随着土壤深度的变化,土壤中细菌种群结构和数量也会发生改变[20]。本研究中,农田土壤柱状样品中的变形菌门和酸杆菌门的相对丰度随土壤深度增加呈现出独特的变化趋势。酸杆菌门的相对丰度随土壤深度增加逐渐增加,而变形菌门则相反。这是由于变形菌门大多由异养细菌构成,适合在C、N含量丰富的环境中生存[21],而酸杆菌门被认为是寡营养细菌,主要生存在营养不良且有机碳浓度较低的酸性环境中[22]。本研究的土壤理化参数结果显示,随土壤深度的增加,NH+4-N和TOC含量减少,故变形菌门和酸杆菌门相对丰度随土壤深度呈相反趋势。
Griffiths等[23]对英国各地超过1 000个土壤剖面样品中细菌群落的多尺度空间分布进行了研究,发现pH值与酸杆菌门丰度呈反比,Rousk等[24]的研究也得出类似结论。本研究结果显示,酸杆菌门的丰度随着pH值的增加而增加。土壤pH是影响酸杆菌丰度和多样性的重要环境因子,但其他环境因素不可忽视。本研究中,pH值随土壤深度的增加呈递增趋势,但NH+4-N、TOC和TKN的含量则随之降低,形成了相对寡营养的环境,为寡营养细菌酸杆菌门营造了适宜的生存环境。Barns等[25]研究发现,大量酸杆菌门亚群在低pH环境中不适宜生存,也会导致酸杆菌门总丰度减少,Jones等[26]的研究结果也证实了该发现。在属水平上,节杆菌属是GJ的主要优势菌属。节杆菌被认为是专性好氧菌,植物根系的泌氧作用促进了节杆菌在小麦根际土壤中的富集。另一方面,隶属于酸杆菌门的亚群Gp6、Gp4和Gp1的相对丰度随pH值呈现出不同趋势,其中Gp6和Gp4的相对丰度随pH值呈现正相关趋势,而Gp1相对丰度随pH值呈现负相关趋势,这与Turlapati等[27]对30个土壤样品中的酸杆菌亚群的研究结果相似。
共现网络分析揭示了细菌之间的相互作用,能够对细菌结构和组装模式进行全面的了解。其中,高度相关的基础单元分类群对细菌群落的功能起着关键作用[28]。本研究表明,在具有紧密联系的不同微小生境中,细菌组成以及细菌间的相互作用均具有独特性。例如,在Module 1中存在大量的变形菌门和少量的酸杆菌门和放线菌门细菌,其中伯克霍尔德菌属(Burkholderia),戴氏菌属(Dyella),马赛菌属(Massilia)在Module 1中起着关键作用。伯克霍尔德菌属因其具有生物防治效果并且能够促进植物生长而被大量报道[29]。戴氏菌属为兼性厌氧细菌,能够将硝酸盐还原成亚硝酸盐,促进反硝化进程[30]。马赛菌属广泛存在于植物根际环境中,能够增强植物抗逆性,还具有降解多环芳烃的功能[31]。在Module 2中,酸杆菌门占据较高的丰度,特别是Gp6和Gp4两个亚群,Navarrete等[32]的研究也得出类似的结论。此外,本研究发现,在Module 1与Module 2交界处存在大量的显著负相关,而这两个Module内部则存在大量的显著正相关,表明Module 1与Module 2之间可能存在着竞争关系,而在各自内部各节点之间存在紧密的合作关系。
4 结论
(1)对农田不同深度土壤(0≤FG-A≤5 cm、5<FG-B≤10 cm、10<FG-C≤15 cm、15<FG-D≤20 cm)及小麦根际土壤(GJ)的细菌群落结构进行了研究。在门水平上,变形菌门、酸杆菌门、放线菌门和厚壁菌门为优势菌门;在属水平上,节杆菌属在小麦根际土壤中占据优势,Gp6则在柱状土壤中占据优势。
(2)酸杆菌门相对丰度随土壤pH值的增加而增加。RDA分析结果表明,TKN、TOC、NH+4-N和pH是影响细菌群落结构特征差异的关键环境因子。此外,基于OTU的样本聚类树分析,表明GJ、FG-A和FG-B相互聚类。
(3)共现网络分析表明,不同模块的细菌组成以及细菌间的相互作用均有所差异。Module 1和Module 2内部各节点之间存在大量合作关系,但Module 1与Module 2之间处于竞争状态。
