RP-HPLC 梯度洗脱法测定盐酸金霉素有关物质方法研究
2024-01-10肖钦钦张银花裴昆陈希段和祥章红江
肖钦钦,张银花,裴昆,陈希,段和祥,章红江
西省药品检验检测研究院,国家药品监督管理局中成药质量评价重点实验室,江西省药品与医疗器械质量工程技术研究中心,江西 南昌 330029
金霉素(chlortetracycline)为四环素类抗生素,被广泛应用于多种细菌感染疾病的治疗,对革兰阳性、阴性细菌和衣原体、支原体、立克次体均有较强的抑制作用[1]。金霉素是人类从金色链霉菌中发酵得到的第一个天然四环素类抗生素[2-3],该类抗生素因化学结构含有许多羟基、烯醇羟基及羰基导致其在酸性、碱性、中性、氧化等条件下均可降解形成多种降解杂质[4-5],金霉素及其已知杂质的化学结构式如图1 所示。金霉素属于两性化合物,等电点为pH 7.4,不易溶于水,能与各种酸、碱形成盐,最常见的为盐酸盐。从国家药品监督管理局网站查询可知,盐酸金霉素原料药现有生产企业2 家,批文2 个,但仅一家企业仍在生产,其制剂有眼膏和软膏2 种剂型,其中盐酸金霉素眼膏占主要市场,临床用于治疗细菌性结膜炎、睑腺炎、细菌性眼睑炎及沙眼,但其质量状况总体评价有待提高[6]。

图1 金霉素和已知杂质的化学结构式
盐酸金霉素现行质量标准《中国药典》2020 年版[7]有关物质检查方法无法有效控制产品质量,该方法采用等度洗脱液相色谱法,部分杂质无法洗脱出来,且杂质计算方法不合理,因此,盐酸金霉素现行质量标准中有关物质测定方法亟待改善[8]。目前,国外药典仅《英国药典》(BP)[9]、《欧洲药典》(EP)[10]收载了盐酸金霉素有关物质检查方法,以不同比例的高氯酸-二甲基亚砜-水体系作为流动相,采用超高效液相色谱(UPLC)进行梯度洗脱,除控制盐酸四环素、4-差向金霉素(杂质A)两个杂质外,还将杂质B、D、E、G、H、J、K、L 作为特定杂质分别予以控制。本研究参照BP 和EP 方法,采用高效液相色谱法,建立有关物质梯度洗脱法,以便更准确更有效地分析盐酸金霉素原料药的杂质状况,从而为盐酸金霉素制剂的质量控制提供保障。
1 仪器与试药
1.1 仪器
MS 205DU 电子天平(德国梅特勒公司),LC-20AT 高效液相色谱仪(日本岛津公司),LC-20AD高效液相色谱仪(日本岛津公司)。
1.2 试药
盐酸金霉素原料药,共13 批,均为同一生产企业提供。对照品:盐酸金霉素(批号:130489-201403,含量:94.1%),盐酸四环素(批号:130488-201604,含量:98.4%),4-差向金霉素(批号:130404-201210,含量:90.1%),杂质D(4-差向四环素,批号:130401-200209,供系统适用性用),杂质I(脱水差向四环素,批号:130403-201205,含量:81.5%),均购于中国食品药品检定研究院。杂质E(批号:D230730,含量:90.0%),杂质B(批号:D0300000,含量:92.4%),杂质G(批号:1790000,含 量:96%),杂 质J(批 号:A1200000,含 量:93.0%),杂质K(批号:E578060,含量:95%),杂质L(批号:A637740,含量:90.39%),金霉素系统适用性对照品(批号:Y0001451,供系统适用性试验用,包含杂质A、B、D、E、G、H、J、K、L),均为EP 对照品。
二甲基亚砜(TEDIA 公司,色谱纯),甲醇、乙腈(Sigma-Aldrich 公司,色谱纯),水为超纯水,高氯酸、盐酸等均为分析纯。
2 方法与结果
2.1 色谱条件
采用Ultimate XB-C8 色谱柱(4.6 mm×250 mm,5 μm);流动相A为高氯酸-水-二甲基亚砜(16∶934∶525)(pH<2.0),流动相B 为乙腈,流动相C 为甲醇,线性梯度洗脱;流速为0.8 mL/min;柱温为45 ℃;检测波长为280 nm;进样量为20 μL。梯度洗脱程序见表1。
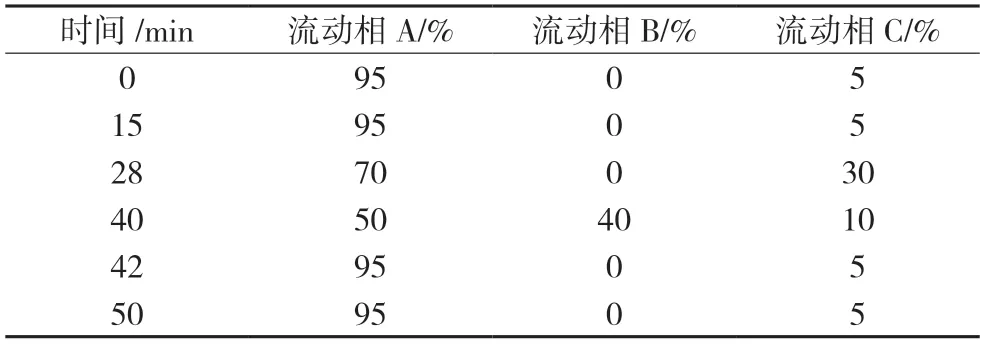
表1 梯度洗脱程序
2.2 系统适用性试验
取盐酸金霉素对照品约10 mg,精密称定,置10 mL 量瓶中,加0.01 mol/L 盐酸溶液溶解并稀释至刻度,摇匀,于80 ℃水浴中加热30 min,放冷。精密量取该溶液20 μL,注入液相色谱仪,记录色谱图,结果各杂质色谱峰之间均能有效分离,分离度均大于1.5,见图2。
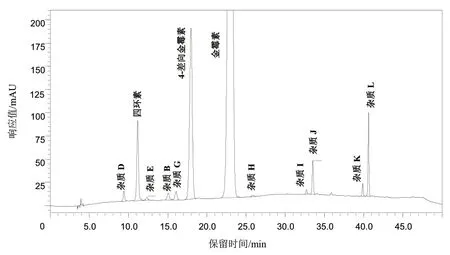
图2 系统适用性试验色谱图
2.3 溶液制备
2.3.1 稀释剂的制备 取盐酸0.9 mL,加水稀释至1 000 mL,摇匀,即得。
2.3.2 供试品溶液的制备 取盐酸金霉素原料药约50 mg,精密称定,置50 mL 量瓶中,加稀释剂溶解并稀释至刻度,摇匀。
2.3.3 对照品溶液(1)的制备 取盐酸四环素对照品、4-差向金霉素对照品各适量,精密称定,加稀释剂溶解并定量稀释制成每1 mL 中分别含80 μg 与40 μg 的溶液。
2.3.4 对照品溶液(2)的制备 取盐酸金霉素对照品适量,精密称定,加稀释剂溶解并定量稀释制成每1 mL 中含盐酸金霉素10 μg 的溶液。
2.3.5 灵敏度溶液的制备 精密量取对照品溶液(2)适量,用稀释剂定量稀释制成每1 mL 中含0.5 μg的溶液。
2.4 方法学验证
2.4.1 线性关系考察 取盐酸金霉素对照品适量,精密称定,加稀释剂溶解制成含盐酸金霉素1 000、800、500、250、125、62、31 μg/mL 的系列标准曲线溶液;取盐酸四环素对照品适量,精密称定,加稀释剂溶解制成每1mL 中约含盐酸四环素200、100、50、25、12、6、3 μg/mL 的系列标准曲线溶液;取4-差向金霉素对照品适量,精密称定,加稀释剂溶解制成每1 mL 中约含4-差向金霉素160、80、40、20、10、5、2 μg/mL 的系列标准曲线溶液。分别精密量取上述3 个成分的标准曲线溶液各20 μL,按“2.1”项下色谱条件进行测定,记录色谱图。各成分以峰面积为纵坐标(Y),质量浓度为横坐标(X),分别进行回归分析。结果显示,3 个成分的标准曲线在各自浓度范围内均呈线性,见表2。

表2 线性关系考察结果
2.4.2 检测限 取“2.4.1”项下各成分最低浓度水平线性溶液逐步稀释,按“2.1”项下色谱条件进行液相色谱分析,以信噪比S/N ≥3 为检测限,盐酸金霉素检测限为0.033 5 μg/mL,盐酸四环素检测限为0.077 9 μg/mL,4-差向金霉素检测限为0.155 6 μg/mL。
2.4.3 重复性 取盐酸金霉素原料药,按“2.3.2”项下方法制备供试品溶液6 份,按“2.1”项下色谱条件进行液相色谱分析,计算各杂质的百分含量,结果6 份供试品中各杂质含量基本无变化,RSD 均小于3.5%,见表3。说明该方法重复性较好。
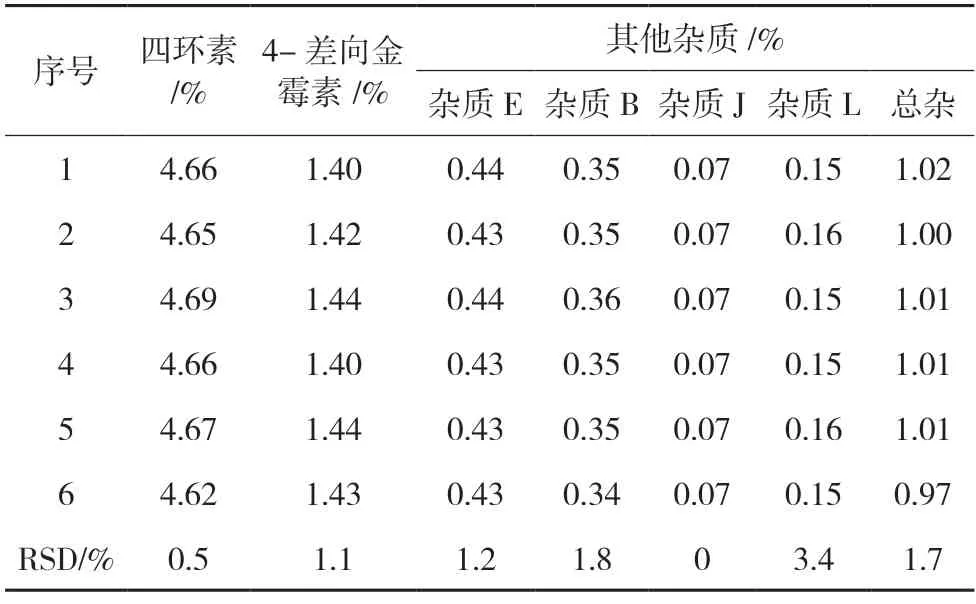
表3 重复性试验结果
2.4.4 准确度 取已知杂质含量的供试品6 份,各约25 mg,精密称定,分别加入对照品溶液(1)适量,按“2.3.2”项下方法制备测试溶液,精密量取测试溶液各20 μL,注入液相色谱仪,记录峰面积,计算回收率。2 种杂质回收率RSD 均小于1.5%,见表4,表明该方法的准确度较好。
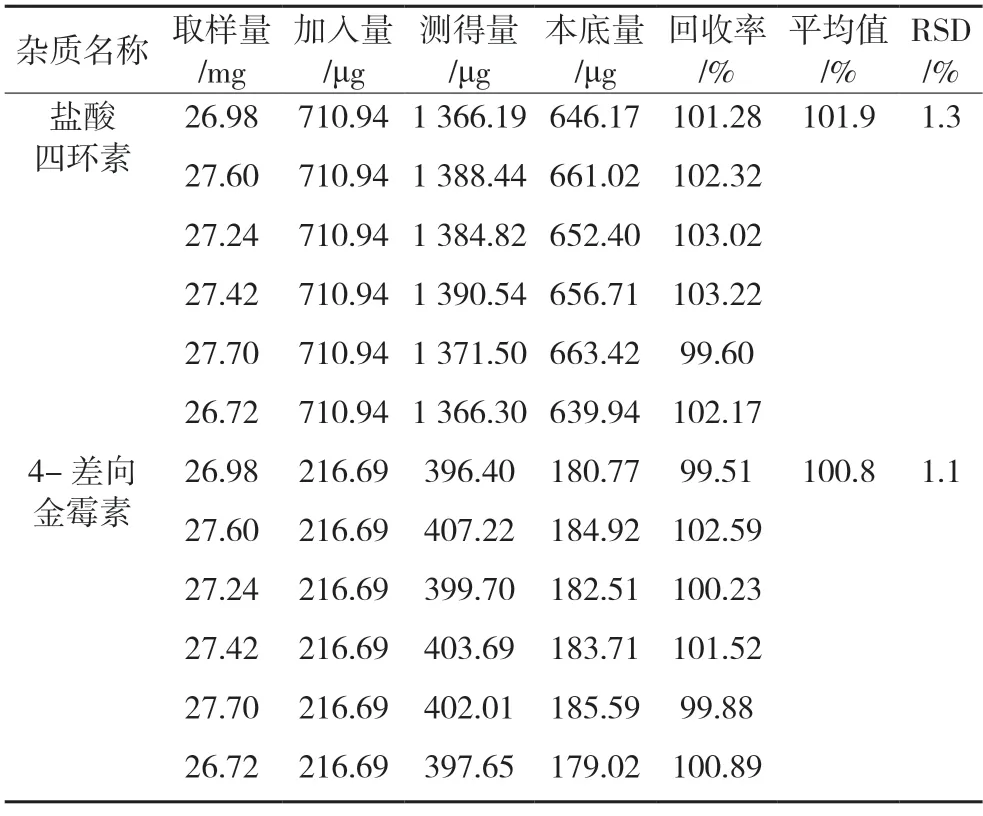
表4 回收率试验结果
2.4.5 专属性 取供试品溶液分别进行以下试验:(A)高温降解,取供试品溶液5 mL,置100 ℃水浴中加热15 min,冷却;(B)光降解,取供试品溶液5 mL,在3 500 lx 光照强度下放置20 h;(C)碱破坏,取供试品溶液5 mL,加0.1 mol/L 氢氧化钠溶液1 mL,室温放置5 min 后,加0.1 mol/L 盐酸溶液1 mL,摇匀;(D)酸降解,取供试品溶液5 mL,加10 mol/L 盐酸溶液2 mL,室温放置30 min 后,加10 mol/L 氢氧化钠溶液2 mL,摇匀;(E)氧化降解,取供试品溶液5 mL,加30%过氧化氢溶液2 mL,摇匀,室温放置2 d。吸取上述5 种降解溶液各20 μL,分别注入液相色谱仪,记录色谱图。
结果显示,盐酸金霉素对高温、碱、酸、氧化非常敏感。高温条件下主要降解为4-差向金霉素、杂质J、杂质L;酸性条件下主要降解为杂质G、杂质J、杂质L;碱性条件下主要降解为杂质G;氧化条件下主要降解为杂质H。盐酸金霉素经强制降解后,各杂质峰与主峰之间均能有效分离,表明方法的专属性强。
2.4.6 稳定性试验 取供试品溶液在室温下放置,分别于不同时间(0、1、2、4、6、8、10、12 h)进样分析,计算各杂质百分含量。结果显示供试品溶液中4-差向金霉素含量变化RSD 为31.4%,增加约1.4倍;杂质J 和杂质L 含量呈增加趋势;其他杂质含量变化均较小。说明供试品溶液不稳定,需临用新制。
2.5 样品测定
按“2.3”项下方法制备盐酸金霉素供试品溶液、对照品溶液、灵敏度溶液,按“2.1”项下色谱条件对13 批盐酸金霉素原料药进行有关物质检查,结果均检出盐酸四环素、4-差向金霉素、杂质B、杂质E、杂质J、杂质L,未检出杂质D、G、H、I、K 及其他未知杂质。除盐酸四环素含量为4.1%~5.2%,4-差向金霉素含量为0.9%~2.3%外,其他杂质总量为0.6%~1.0%,其中杂质B 含量为0.1%~0.4%,杂质E 含量为0.3%~0.6%,杂质J 含量为0.03%~0.07%,杂质L 含量为0.06%~0.15%,13 批原料药批间差异较大,但均在拟定限度范围内。
3 讨论
3.1 流动相及系统适用性试验的确定
通过降低流动相中二甲基亚砜比例,并调节甲醇、乙腈比例进行梯度洗脱的方法,可明显解决现行质量标准中高比例的二甲基亚砜易致系统超压且色谱峰保留时间漂移大、等度洗脱难以将杂质完全洗脱出来以致杂质残留加快色谱柱柱效降低等问题。
利用盐酸金霉素对照品进行热降解,采用单个杂质对照品对各杂质峰进行定位,结果显示该降解溶液色谱图中主要杂质峰与BP 或EP 中系统适用性对照品中杂质峰一致,盐酸金霉素及其11 个已知杂质均能有效分离,确定了系统适用性试验方法及要求。
3.2 各杂质计算方法及限度的确定
BP 或EP 收载的盐酸金霉素原料药有关物质杂质计算方法,盐酸四环素采用外标法计算不得过6.0%;杂质G、J、K、L 采用加校正因子(依次为1.4、0.3、0.4、0.4)主成分外标法计算,依次不得过0.2%、0.3%、0.15%、0.2%;杂质A、B、D、E、H 均采用不加校正因子主成分外标法计算,依次不得过4.0%、1.0%、0.2%、1.0%、0.2%,其他单个杂质不得过0.10%,杂质抛弃限为0.05%,各杂质总和(除盐酸四环素、杂质A 外)不得过2.0%。
对原料药中检出的杂质J、L 按现行质量标准方法计算含量分别为0.1%~0.3%、0.2%~0.5%,平均含量为0.2%、0.3%,杂质J 含量大于0.2%的样品率为38.5%,杂质L 含量超出BP 或EP 限度样品率为92.3%,显然该计算方法不合理。本研究考察了杂质J、K、L 的相对校正因子,结果差异甚小,因此相对校正因子及限度均参照BP 和EP;杂质G 在原料药及制剂中均未检出,不作为特定杂质进行控制;其他杂质除杂质E 限度一致外,杂质B(不得过0.5%)和杂质D、H、I、K(均不得过0.1%)限度均低于BP 或EP 标准;盐酸四环素和杂质A 计算方法与限度仍参照现行质量标准,以更严格的标准控制国内盐酸金霉素原料药的质量。
