联吡啶二酰胺配体对U(VI)/Mo(VI)的萃取分离及机理
2024-01-08修涛元张萌叶国安矫彩山袁立永石伟群
修涛元, 张萌, 叶国安,3, 矫彩山, 袁立永, 石伟群
(1.哈尔滨工程大学 核科学与技术学院,黑龙江 哈尔滨 150001; 2.中国科学院 高能物理研究所, 北京 100049; 3.中国原子能科学研究院, 北京 102413)
医用放射性同位素在疾病的诊断和治疗中发挥着非常重要的作用,其中99mTc药物被广泛应用于疾病诊断[1-3]。99mTc主要由其母核素99Mo通过99Mo/99mTc发生器制备[4-5]。目前,99Mo主要获取方式有反应堆生产、加速器制备和中子发生器制备等[6-7]。其中利用反应堆中高浓缩铀/低浓缩铀靶体的裂变产生99Mo是最主要的方式[8-9]。制备后靶件溶解液中含有大量铀和裂片元素,其中U(VI)和Mo(VI)化学性质相似,这使得U(VI)/Mo(VI)分离成为一个难题。因此,研究U(VI)/Mo(VI)分离具有科学价值和现实重要意义。
U(VI)/Mo(VI)的分离方法有萃取法、离子交换法、沉淀法、吸附法和挥发法等[10]。溶剂萃取因其条件温和、操作简单、易于产业化而广泛应用于核燃料循环前后端[11-12]。Lsasheen等[13]将LIX 622N用于从含有Mo(VI)和U(VI)的硫酸盐浸出液中萃取回收Mo(VI),并获得高纯度的MoO3(99.99%)。Behera等[14]用Alamine 310、TBP、DPSO及其苯中的混合物从磷酸水溶液对铀钼分离进行研究,Alamine 310、TBP和DPSO可以实现分离,其中DPSO在8 mol/L H3PO4下,U(VI)/Mo(VI)分离因子最大可达到180.37。Behera等[15]用有机磷酸、次膦酸及其硫代衍生物在 0.1~1.0 mol/L 的HCl下萃取分离U(VI)/Mo(VI),比较了 PC-88A、Cyanex 272、Cyanex 301和Cyanex 302的萃取分离U(VI)/Mo(VI)效果,Cyanex 301在1 mol/L的HCl 下可以实现U(VI)/Mo(VI)分离。许多配体设计出,并用于分离U(VI)/Mo(VI),但大多数配体含有磷,不符合二次废物最小化的“CHON”原则[16]。氮杂环酰胺类配体用于萃取分离锕系元素。其中具有代表性的有吡啶二酰胺类、联吡啶二酰胺类和邻菲啰啉二酰胺类等。该类配体在酸性和辐照条件具有稳定性好、动力学快和合成简单等特点。同时,这类配体又符合“CHON”原则,因此得到广泛关注和研究。其中联吡啶二酰胺类配体自身柔性较大,可以通过自身的旋转更好地与金属离子配位。目前,联吡啶二酰胺配体在锕系元素分离中的潜力越来越受到人们的关注。该类配体没有相关文献进行U(VI)/Mo(VI)分离研究。
本文采用3种不同烷基取代基的联吡啶二酰胺配体(Et-EB-DABP、But-EB-DABP和Oct-EB-DABP)作为萃取剂。利用溶剂萃取法,在不同稀释剂、酸度、时间、配体浓度和U(VI)浓度等条件下,对U(VI)/Mo(VI)分离进行系统研究;选用H2O、HNO3(0.01 mol/L)和Na2CO3(5%) 3种反萃剂进行反萃实验;通过傅里叶变换红外光谱(FT-IR)和电喷雾电离质谱(ESI-MS)进行配位机理研究。
1 配体合成及实验方法
1.1 配体合成
配体合成路线如图1所示。将化合物1 (4.69 g,0.025 mol)和KMnO4(26.2 g,0.17 mol)加入到300 mL水中;将混合物加热至90 ℃搅拌14 h;冷却至室温后过滤,将滤液用Et2O萃取3次;所得水溶液通过1 mol/L的HCl调至pH为2,过滤干燥后的得到白色化合物2[17]。将SOCl2(25 mL)加入装有化合物2 (1.00 g,4.10 mmol)的烧瓶里,85 ℃回流4 h;通过旋蒸除去多余的SOCl2,得到黄色化合物3[18];化合物3溶于CH2Cl2(25 ml)中,将NEt3(1.66 g,16.40 mmol)加入到CH2Cl2(10 ml)并分别加入化合物4 (1.74 g, 9.02 mmol)、5 (2.00 g, 9.02 mmol)或6 (2.50 g, 9.02 mmol)[19],化合物4、5和6合成方法参照文献[20],在冷CH4CH2OH浴下缓慢加入到化合物3的CH2Cl2溶液中;在40 ℃下回流4 h后,通过旋蒸除去溶剂,用硅胶柱层析法纯化(展开剂: CH3OH/CH2Cl2=1/20),得到3种白色联吡啶二酰胺配体:化合物7 (Et-EB-DABP(L1))、化合物8 (But-EB-DABP(L2))和化合物9 (Oct-EB-DABP(L3))。3种配体已经NMR (1H,13C) 和ESI-MS确认,如图2所示。

图1 配体合成路线Fig.1 The synthetic route of the ligands

图2 3种配体(L1, L2和L3)的NMR(1H, 13C)和ESI-MS谱图Fig.2 The NMR(1H, 13C) and ESI-MS spectra of three ligands (L1, L2, and L3)
1.2 萃取及光谱实验方法
将一定量UO2(NO3)2·6H2O和Na2MoO4分别溶于1 mol/L的HNO3并用容量瓶定容,制备U(VI)和Mo(VI)母液。根据不同实验条件配置不同水相。将一定量的L1、L2和L3分别溶于不同稀释剂得到有机相。预平衡有机相,将有机相与水相混合,在室温下充分振荡30 min, 相比(O/A)始终为1,进行萃取和反萃。经7 000 r/min离心分离3 min后,取水相,用电感耦合等离子体发射光谱仪(ICP-OES)测定U(VI)和Mo(VI)的浓度。有机相U(VI)和Mo(VI)的浓度通过差减法得到。分配比D和分离因子β为[21-22]:
D=[M]org/[M]aq
(1)
βU/Mo=DU/DMo
(2)
式中:[M]org和[M]aq分别表示萃取后有机相和水相中的金属离子浓度。
反萃实验选用Na2CO3(5%)、H2O和HNO3(0.01 mol/L)作为反萃剂。将萃取后有机相与反萃剂等体积混合,离心后测定水相中金属离子的浓度。反萃率S为[20]:

(3)
式中:[M]str.aq和[M]ext.aq分别为反萃后水相中的金属离子浓度和反萃前有机相中的金属离子浓度。
萃取前后FT-IR通过的将L1、L2和L3萃取U(VI)前后的有机相采用溴化钾压片法在500~2 000 cm-1波长进测试。取一定量的L1、L2和L3分别溶于乙腈中配置浓度为0.01 mmol/L的母液。取1 mL配体乙腈溶液并逐次加入2.5 μL的1 mmol/L U(VI)乙腈溶液调节U(VI)与配体比例。将上述溶液置于恒温振荡器中震荡30 min,以确保配位平衡。随后,对溶液进行ESI-MS测试。
2 萃取结果与机理分析
2.1 稀释剂的影响
为了研究配体在不同溶剂中的萃取性能,选用5种不同稀释剂(正辛醇、甲苯、二氯甲烷、1,2-二氯乙烷和间硝基三氟甲苯)进行萃取实验。萃取条件为:[L1]=[L2]=[L3]=2 mmol/L、[HNO3]=4.00 mol/L、[U(VI)]=0.5 mmol/L、t=30 min、T=298 K和O/A=1。不同稀释剂的影响图3所示,3种配体对U(VI)具有较好的萃取效果,其分配比由低到高依次为:正辛醇<甲苯<二氯甲烷<1,2-二氯乙烷<间硝基三氟甲苯。由于间硝基三氟甲苯对联吡啶二酰胺类配体具有良好的溶解度,同时具有良好的疏水性和较高介电常数,可提高萃取能力[20]。因此,选择间硝基三氟甲苯作为后续实验的稀释剂。
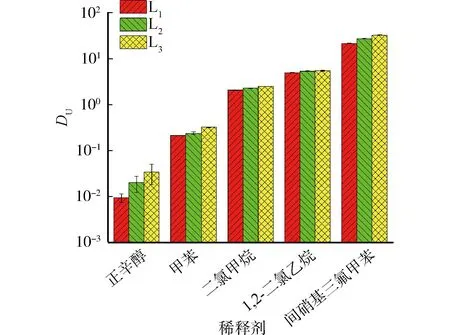
图3 稀释剂对3种配体(L1, L2和L3)萃取U(VI)和Mo(VI)的影响Fig.3 Influence of diluents on the DU value by L1, L2 and L3 ligands
2.2 HNO3浓度的影响
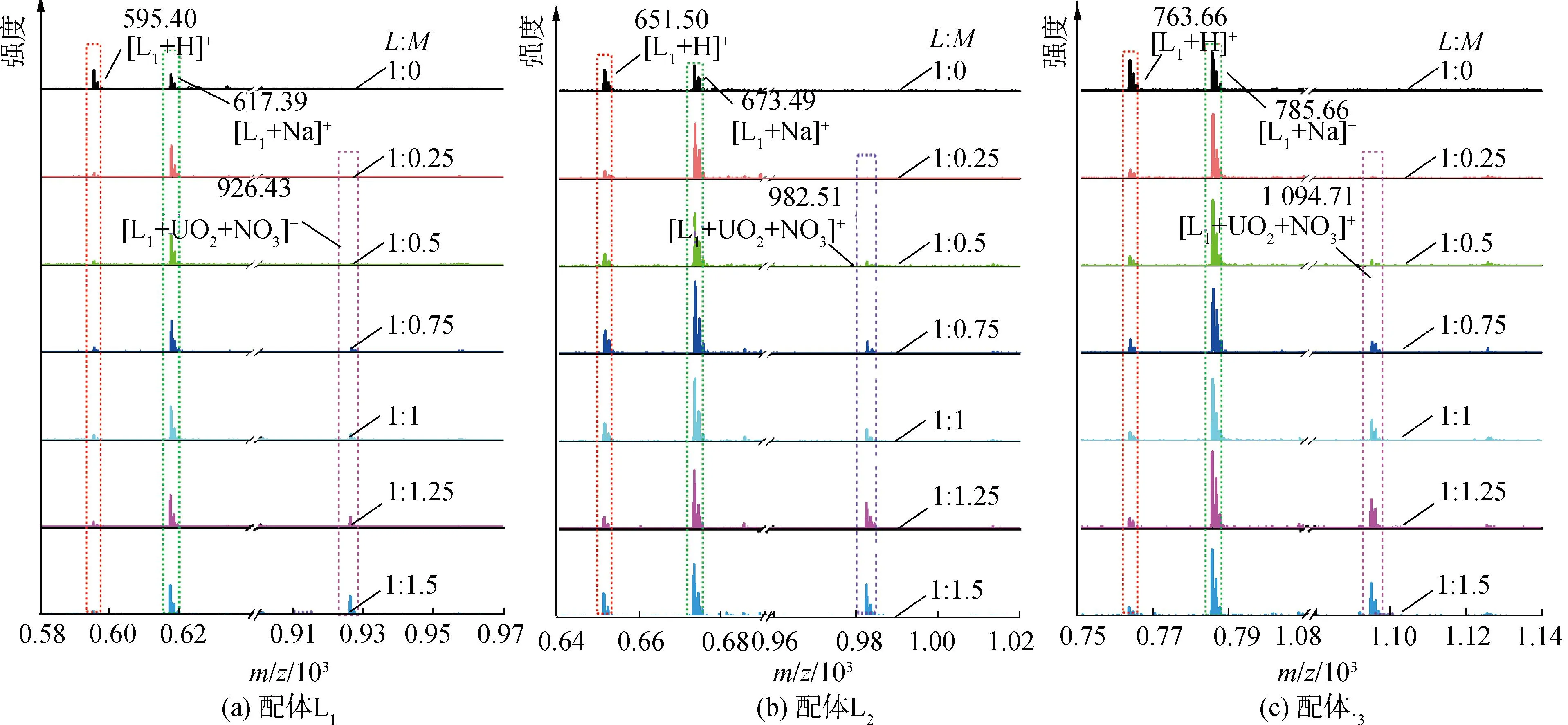
研究HNO3浓度的影响时,萃取条件为:[L1]=[L2]=[L3]=2 mmol/L、0.1 mol/L≤[HNO3]≤4 mol/L、[U(VI)]=0.5 mmol/L、[Mo(VI)]=0.2 mmol/L、t=30 min、T=298 K和O/A=1。DU、DMo和βU/Mo随HNO3浓度变化如图4所示。3种配体对U(VI)和Mo(VI)具有较强的分离能力,分离能力由大到小依次为L1 图4 HNO3浓度对3种配体(L1, L2和L3)萃取U(VI)和Mo(VI)的影响Fig.4 Influence of HNO3 concentration on the D value and β value of U(VI) and Mo(VI) by L1, L2 and L3 ligands 萃取动力学是评价萃取性能的重要参数之一。研究接触时间的影响时,萃取条件为:[L1]=[L2]=[L3]=2 mmol/L、[HNO3]=0.5 mol/L、[U(VI)]=0.5 mmol/L、[Mo(VI)]=0.2 mmol/L、1 min≤t≤30 min、T=298 K和O/A=1。接触时间对U(VI)和Mo(VI)萃取的影响如图5所示。随着接触时间的增加,Mo(VI)的分配比一直趋于零。L1、L2和L3对U(VI)的萃取在15 min内达到平衡。3种配体的U(VI)萃取动力学速度较快。与邻菲啰啉酰胺类配体对比,动力学相对较慢,原因为联吡啶骨架在萃取过程中,需要扭曲自身,从而需要更长的时间才能达到平衡[20]。 图5 时间对3种配体(L1, L2和L3)萃取U(VI)和Mo(VI)的影响Fig.5 Influence of contact time on the D value of U(VI) and Mo(VI) by L1, L2 and L3 ligands 研究配体浓度的影响时,萃取条件为:2 mmol/L≤[L1]=[L2]=[L3]≤10 mmol/L、[HNO3]=0.5 mol/L、[U(VI)]=0.5 mmol/L、[Mo(VI)]=0.2 mmol/L、t=30 min、T=298 K和O/A=1。如图6所示,随着配体浓度的增加,配体对U(VI)的分配比迅速增加,而对Mo(VI)的分配比一直趋近于零。斜率分析法通常用于确定配体与金属离子的化学计量比。由于L1、L2和L3对Mo(VI)的萃取效果较差,斜率分析法无法分析配体与Mo(VI)的化学计量比。因此,只研究了配体与U(VI)的化学计量比。在溶液中,萃取平衡等式为[22]: 图6 不同浓度萃取U(VI)和Mo(VI)的影响Fig.6 Influence of ligand concentration on the D value of U(VI) and Mo(VI) 萃取平衡常数KexU和U(VI)的萃取分配DU为: 将式(4)~(6)重新组合得到: > (4) (5) (6) (7) 以分配比的对数值作为纵坐标,以萃取剂浓度的对数值作为横坐标,可得到线性等式的斜率值为金属离子与萃取剂分子形成配合物的化学计量比。如图6所示,L1、L2和L3与U(VI)的斜率分别为0.58、0.56和0.53,说明在间硝基三氟甲苯溶剂中,L1、L2和L3与U(VI)在萃取过程中的化学计量比为1∶1,形成的配合物为:UO2(NO3)2·L。由于L1、L2和L3为四齿配体,易于占据铀酰平面六配位中的4个位点,而其自身垂直方向存在2个氧原子阻止配体进一步配位,因此不能形成配体与铀酰比例为2∶1配合物。 配体对金属离子的萃取容量是衡量其萃取性能的重要指标。为了探究3种配体(L1、L2和L3)对U(VI)的萃取容量。将配体浓度设为2 mmol/L,通过改变U(VI)浓度探究其萃取容量。具体萃取条件为:[L1]=[L2]=[L3]=2 mmol/L、[HNO3]=4.0 mol/L、0.5 mmol/L≤[U(VI)]≤5.0 mmol/L、t=30 min、T=298 K和O/A=1。由图7可知,当U(VI)浓度为5 mmol/L时,有机相中配体与U(VI)的比值接近于1。这表明3种配体对铀的萃取容量均达到理论值[23]。 图7 U(VI)浓度的影响Fig.7 Influence of U(VI) concentration 对于一种具有应用前景的配体,不仅要有良好的萃取性能,同时也有良好的反萃性能。选用HNO3(0.01 mol/L)、H2O和Na2CO3(5%)作为反萃剂。U(VI)的3级反萃如图8所示。3种反萃剂的对U(VI)的反萃效果由高到低:Na2CO3(5%)>H2O>HNO3(0.01 mol/L)。用5% Na2CO3溶液,只需一步就可以达到接近100%的反萃率。这是由于与碳酸盐相比,配体和U(VI)配合物的logβ相对较低[22]。因此,有机相中负载的U(VI)很容易被碳酸盐剥离,从而形成更稳定的UO2(CO3)n[22]。3种反萃的3次反萃率均接近100%,表明配体具有良好的反萃性能。 图8 3种不同反萃剂H2O、HNO3 (0.01 mol/L)和Na2CO3 (5%)对U(VI)的反萃Fig.8 Stripping of U(VI) from organic phase by ultrapure water, 0.01 mol/L HNO3 and 5% Na2CO3 图9 L1、L2和L3萃取U(VI)前后的FT-IR谱图Fig.9 The FT-IR spectra of ligands (L1, L2 and L3) and its complex with U(VI) 由图10可知,L1、L2和L3这3种配体与H+结合的峰分别出现质荷比m/z为595.40、651.50和763.66,以及与Na+结合的峰,出现在m/z为617.39、673.49和785.66。随着U(VI)的不断滴加,3种配体的质谱谱图中逐渐出现配体结合一个铀酰一个硝酸根的配合物[L1+UO2+NO3]+(m/z926.43),[L2+UO2+NO3]+(m/z982.51)和[L3+UO2+NO3]+(m/z1 094.71),且U(VI)的比例越高,相应的峰越强。以上分析可知,U(VI)与L1、L2和L3这3种配体形成U(VI)∶L=1∶1的配合物。本文方法与斜率法结果相吻合。 图10 CH3CN体系U(VI)滴定L1、L2和L3的ESI-MS分析Fig.10 ESI-MS titration of ligands (L1, L2 and L3) with U(VI) in CH3CN 1)3种联吡啶二酰胺配体在最优条件下分离因子达到~2 000,实现U(VI)和Mo(VI)之间的高效分离。 2)研究发现配体烷基链越长,在稀释剂中的溶解性越高,因此对U(VI)的萃取能力越强,进而表现出更强U(VI)/Mo(VI)的分离能力。 3)3种配体萃取动力学快,15 min内达到平衡。3种配体对U(VI)的萃取容量甚至达到理论值。3种反萃剂3次反萃百分比都接近100%,表明该类配体具有良好的循环利用潜力。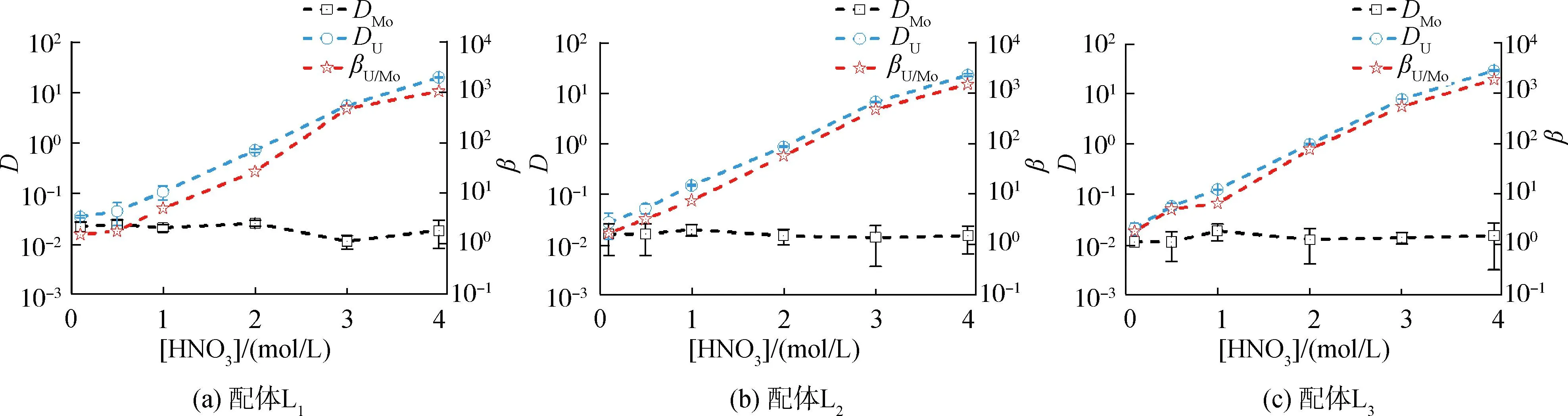
2.3 时间的影响

2.4 配体浓度的影响

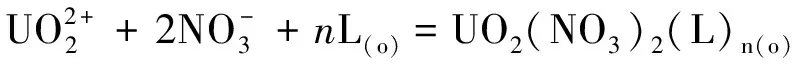
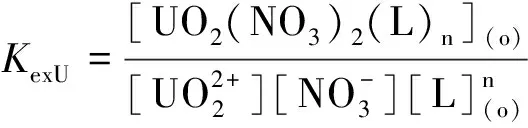


2.5 U(VI)浓度的影响

2.6 反萃

2.7 FT-IR分析


2.8 ESI-MS滴定分析
3 结论
