肝豆状核变性患者临床特征及基因突变位点分析*
2024-01-07袁宇初薛美珠
王 琦,周 峰,袁宇初,薛美珠
肝豆状核变性,又称Wilson病(Wilson’s disease,WD),系铜代谢紊乱性疾病,通常由基因突变导致,发病年龄在3~30岁之间,可累及多个脏器,临床表现十分复杂,症状多样。根据临床表现,该病主要分为肝型、脑型和混合型等[1]。当病变累及肝脏时,临床表现主要为恶心、乏力、黄疸,严重者可以发生肝硬化,甚至发生肝功能衰竭[2]。当病变累及中枢神经系统时,患者常有行为异常、性格改变和共济失调等症状[3]。肝豆状核变性与ATP7B基因密切相关,该基因主要编码P型ATP酶,编码的蛋白质与铜蓝蛋白前体特异性结合,并形成铜蓝蛋白。当机体铜含量过高时, P型ATP酶将过剩的铜元素转移到胆汁中排出,防止机体铜含量过高产生毒性[4]。ATP7B突变的最常见类型为错义突变,P型ATP酶合成大量减少,机体过量的铜元素无法及时被排出体外,积累在肝脏和大脑等重要器官,威胁生命健康[5]。主要根据临床表现、影像学结果、生化检测、家族史和基因检测等诊断肝豆状核变性。对于高度疑似肝豆状核变性但临床特点不典型的患者,为进一步确诊需要检测ATP7B基因突变[6]。本研究分析总结了WD患者的临床特征和ATP7B致病突变情况,现报道如下。
1 资料与方法
1.1 研究对象 2017年1月~2022年12月我院诊治的WD患者79例,男性40例,女性39例;年龄为4~71岁,平均年龄为(20.3±13.9)岁。符合中华医学会发布的肝豆状核变性诊治指南的标准,且符合以下各项的3项以上:(1)有肝病症状或出现神经系统症状;(2)血清铜蓝蛋白<0.2 g/L;(3)24 h尿铜>100 μg;(4)存在角膜K-F环;(5)肝铜(干重)>250μg/g;(6)基因检测结果显示ATP7B基因突变;(7)有相关的家族病史。分型标准为:(1)肝型表现为肝脏损害,主要包括肝酶异常、肝功能不全、肝硬化等;(2)脑型表现为神经系统症状、包括精神障碍、运动障碍、姿势障碍、震颤麻痹等;(3)混合型表现为各种类型的症状组合。排除标准为:(1)合并甲型或乙型肝炎病毒感染;(2)合并酒精性肝炎;(3)合并HIV感染;(4)肝细胞癌或肝移植史;(5)恶病质,出现严重的黄疸、腹水、肝性脑病等;(6)合并严重的其它器官器质性病变。本研究经我院医学伦理审查委员会审查并批准,基因检测经患者或患者监护人的同意,所有患者及其家属均已签署纸质知情同意书。
1.2 血清检测 使用瑞士Roche公司生产的Cobas ISE600型全自动生化分析仪检测血生化指标;取血清,采用高通量基因测序法进行基因检测(华大基因检测中心,中国),应用ATP7B基因数据库比对基因突变位点。

2 结果
2.1 三型WD患者一般资料比较 肝型患者初次发病年龄显著小于脑型和混合型患者,而脑型患者出现K-F的比率显著高于肝型和混合型患者,差异具有统计学意义(P<0.05,表1)。
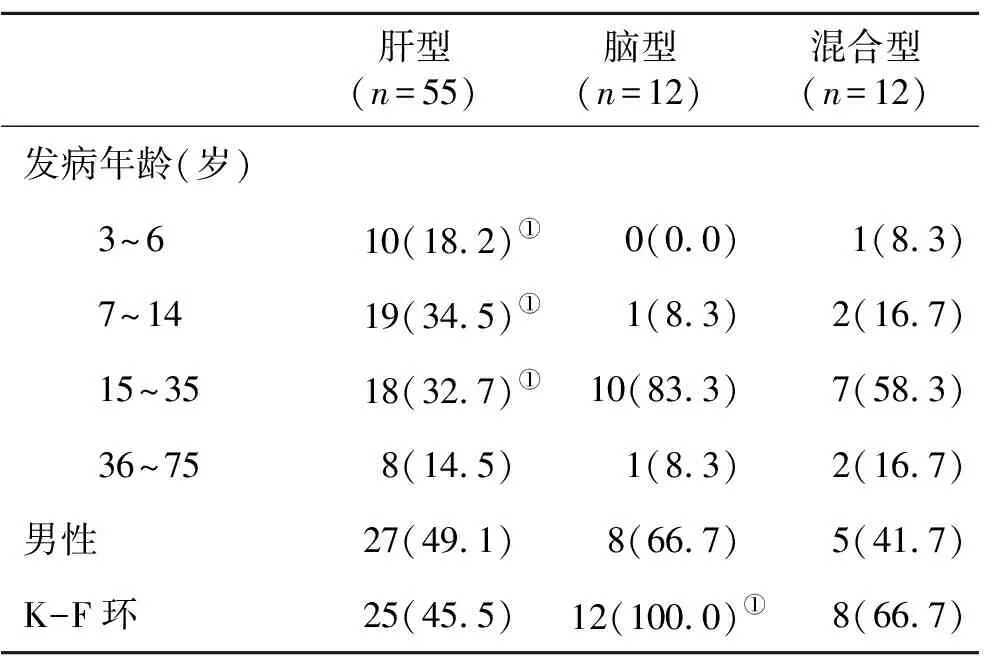
表1 三型WD患者一般资料(%)比较
2.2 三组实验室指标比较 肝型患者血清AST、ALT和铜蓝蛋白水平显著高于脑型或混合型患者,差异具有统计学意义(P<0.05,表2)。

表2 三组实验室指标比较
2.3 三型基因突变位点分析 经高通量基因测序分析,共检出16个基因突变位点,混合型EX11、EX13/CDS13、EX16位点和EX18位点突变比例显著高于其它两型(P<0.05),而肝型EX13和EX8位点突变比例显著高于其它两型(P<0.05,表3)。
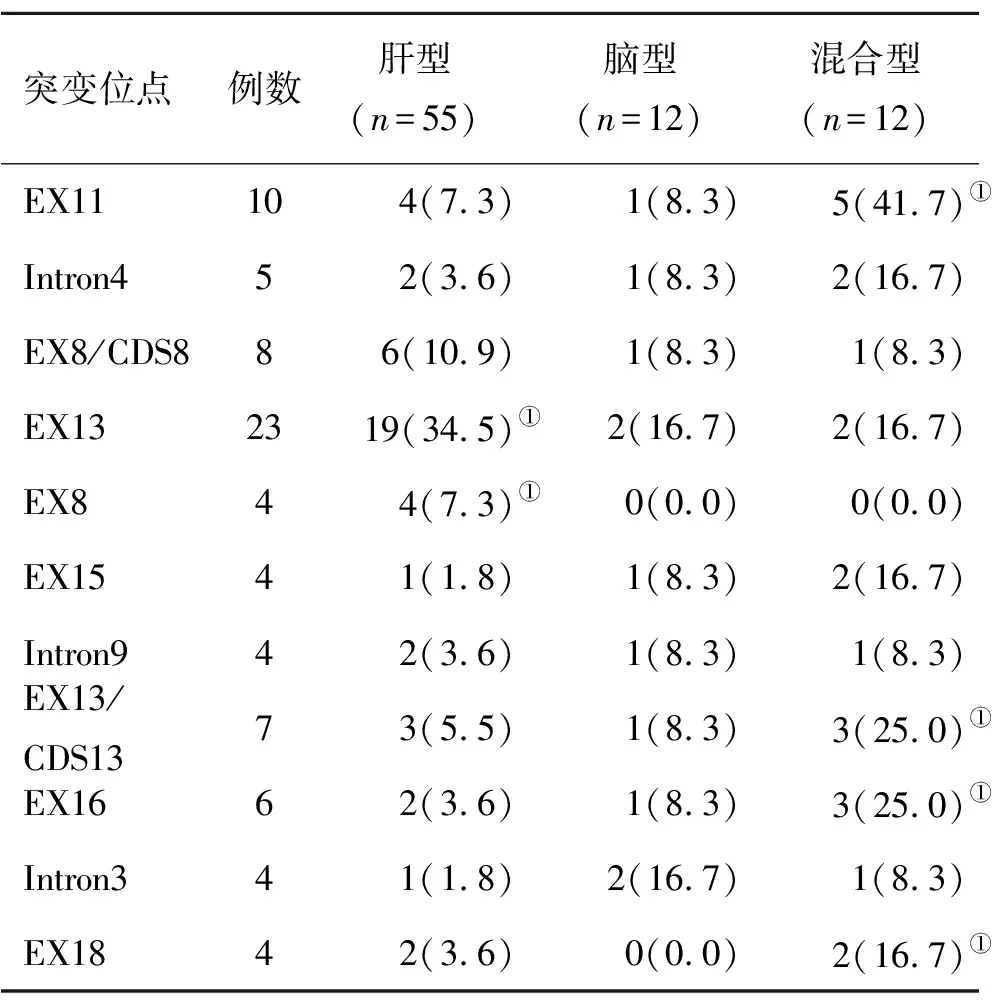
表3 三型基因突变位点分析
3 讨论
肝豆状核变性患者大多数在3~30岁之间起病,发病年龄跨度较大。有研究报道从不满一周岁的婴儿到超过70周岁的老人均可以发病。该病是常染色体隐性遗传病,中枢神经症状往往于肝功能损伤后出现[7]。大脑和肝脏是WD最常累及的部位[8]。中枢神经系统症状和肝脏症状可以相继出现,也可以同时发生[9]。约半数WD在典型临床表现出现前就已经发生了严重的肝病,例如肝硬化,大多数患者在接受肝活检病理学检查时才首次被发现[10]。
不同的临床分型之间发病年龄存在差异[11]。脑型患者平均发病年龄比肝型患者更晚[12]。本研究对各个不同分型的WD患者发病年龄进行了比较,结果发现病变主要累及肝脏,即肝型患者发病年龄较脑型患者更年轻,且初次发病主要为青春期, 而累及中枢神经系统的患者,即脑型患者于青壮年发病的比例更高。本研究对WD患者肝功能进行了评估和分析,不同表型之间血清AST水平和ALT水平差异显著,肝型患者肝脏酶学水平显著高于其它表型患者。在正常情况下,处于生长发育期的青年血清ALP水平可能升高[13]。肝豆状核变性会不同程度损伤下丘脑-垂体-靶腺轴,甲状旁腺激素水平显著降低,导致骨代谢发生异常,出现ALP水平继发性升高[14]。众所周知,K-F环是肝豆状核变性患者典型而突出的眼部特征性表现。有研究表明,与肝型患者相比,脑型患者更容易出现K-F环[15]。通过对所有肝豆状核变性患者K-F环发现率进行分析,我们发现本研究所有脑型患者均出现了K-F环,这与另一项研究结果相一致[16]。铜蓝蛋白的水平可用于诊断肝豆状核变性,但特异性较差。有相关研究表明,10%~30%肝豆状核变性患者铜蓝蛋白水平无异常表现[17]。在我们的研究中,只有少部分患者铜蓝蛋白水平处于正常范围内,大部分患者有不同程度的下降。我们进而分析了肝型、脑型和混合型患者铜蓝蛋白降低是否存在统计学差异。结果显示,三种临床分型患者之间铜蓝蛋白水平存在显著性差异。有研究表明,大约6%WD患者将逐渐进展为肝功能衰竭,后者病情危重且进展十分迅速,治疗反应慢,病死率极高[18]。这些患者具有以下特点:(1)肝功能严重受损;(2)铜蓝蛋白水平极低;(3)发病年龄小;(4)肝脏酶学水平变化主要为AST水平升高为主;(5)尽管肝豆状核变性患病率无显著的性别差异,但是有研究表明,肝豆状核变性的女性患者更易进展为肝功能衰竭。研究报道,部分WD患者还可以合并原发性胆管炎-自身免疫性肝炎重叠综合征或乙型肝炎等严重的肝脏疾病,加重了病情。目前普遍认为肝豆状核变性主要是通过血清过量的铜元素的毒性作用对肝脏造成损伤。研究表明,血清铜元素水平是诊断肝豆状核变性进展为肝功能衰竭的良好指标[19]。血清铜元素水平与患者预后关系密切。铜元素水平越高,患者对治疗的反应越不敏感,预后越差。对于诊断明确的肝功能衰竭患者,应及时使用青霉胺治疗,将失代偿期肝功能转变为代偿期肝病,早期正确诊断肝豆状核变性进展为肝功能衰竭至关重要。
ATP7B突变形式多种多样,主要为错义突变、缺失突变、无义突变和插入突变等。ATP7B 突变主要为几个高频突变,同时可以伴有罕见的位点突变,种族差异十分显著。在欧洲,WD患者H1069Q是最为常见的突变类型,R778L是亚洲WD患者最常见的突变类型。基因诊断是目前诊断WD较为可靠的检查方法,经过高通量检测可以检测到经典的高频突变位点,检测还有助于发现新的突变位点。大约50%WD患者表现为复合型突变。有研究指出,复合突变患者临床表现通常更加严重。本研究未进一步研究疾病进展,如转变为肝功能衰竭的患者,基因突变情况。通过分析基变位点与临床分型之间的关系,我们发现肝型、脑型与混合型患者基因突变位点不同。肝豆状核变性患者基因内含子区突变可能具有致病性,但内含子区基因突变与WD临床表现的严重程度是否具有一定的关系尚不十分明确[20]。在临床诊治WD患者过程中,应当重视对ATP7B内含子区基因突变的研究。
