近红外非富勒烯受体材料在有机太阳电池中的研究进展
2024-01-05曾俊豪谢锐浩
曾俊豪,谢锐浩
(广东技术师范大学光电工程学院,广东 广州 510665)
0 引言
有机太阳电池(Organic Solar Cells,OSCs)被认为是一种环保绿色的太阳能发电技术。与传统的无机光伏相比,有机太阳电池具有重量轻、柔性和半透明等优点,在智慧城市或是室内应用方面有着巨大潜力[1-3]。1986 年,邓青云等[4]用酞菁铜(CuPC)和苝四羧基衍生物制备了第一个双层异质结有机太阳电池,器件效率达到了约1%。自第一个有机太阳电池报道以来,其研究受到了广泛地关注,而且有机太阳电池的能量转换效率(Power Conversion Efficiency,PCE)已从最初的1%迅速发展至现在的20%左右[5]。有机太阳电池的效率突破受益于多方面的科学研究,其中包括给体/受体材料和界面材料的迅速发展,以及器件结构和制备工艺的进步[6-8]。
有机太阳电池的器件结构类似“三明治”,主要由两个电极、界面层和中间的活性层构成(见图1)。其中,活性层由有机半导体材料制备得到,通常含有给体材料和受体材料。有机半导体作为一种活性材料,与硅(Si)、砷化镓(GaAs)和钙钛矿等无机材料相比,有许多不一样的优点,是目前十分有竞争力的光伏材料。其一,有机半导体可以用一个非常薄的活性层(厚度约100 nm)来有效地捕获入射光子,较长的π-π 共轭使其有很高的吸收系数;其二,有机半导体的结构是可以进行修饰的,能够通过精准的化学手段来调节材料的光学性质和电学性质,以及材料的聚集行为。然而,由于有机半导体自身存在的一些问题,使有机太阳电池的器件效率很难提高,即使在p 轨道上电子和空穴可以沿着有机半导体的共轭主链自由移动,但分子间的电荷传输还是比较困难的,所以导致在有机半导体中的载流子迁移率不高。此外,在光致激发后,与无机半导体材料中半径较大的Wannier 激子不同,有机半导体材料中激子主要是半径较小、寿命比较短(扩散距离小于20 nm)的紧密束缚的Frenkel 激子(束缚能有3.1 eV),这需要额外的驱动力来将电子空穴对分离成自由载流子。为了克服这个瓶颈,Heeger 等[9]采用给体材料和受体材料共混,制备了含有给受体材料的本体异质结(Bulk Heterojunction,BHJ)有机太阳电池,大幅度提高了激子的解离效率,提升了光伏性能。
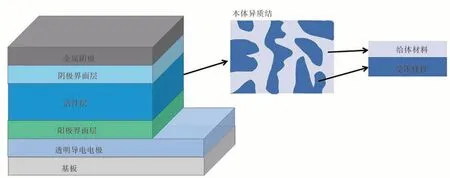
图1 有机太阳电池器件示意图Figure 1 The regular structure of organic solar cells
本体异质结有机太阳电池的出现引起了广泛地关注,有机太阳电池的研究有了突破性的进展,主要是来源于其独特的工作机理。当活性层吸收能量大于能隙的光子时,电子从给体中的最高占据轨道(Highest Occupied Molecular Orbital,HOMO)跃迁至最低空置轨道(Lowest Unoccupied Molecular Orbital,LUMO),形成一种电子-空穴束缚对,即激子(见图2)。随后,激子经过扩散到达给体/受体的界面处,在给受体材料LUMO 或HOMO 能级差驱动下,克服激子中的库仑力束缚,给体材料LUMO能级的电子从而转移到受体材料LUMO 能级,所以给体材料的HOMO 会剩下空穴而受体LUMO 会多出电子,即激子完成解离。解离后的自由的电子载流子、空穴载流子在活性层中传输,分别到达阳极和阴极附近,被电极收集形成光电流。在本体异质结的结构中,共混的给体和受体会形成一个连续的相互贯穿的网络,这不仅大大增加了给体和受体的界面的面积,还有利于激子的解离和电荷的传输,从而提高了器件的光伏性能。总而言之,本体异质结的出现为有机太阳电池的快速发展奠定了坚实的基础。
富勒烯及其衍生物(如PC61BM 和PC71BM)具有较高的电子迁移率和合适的LUMO 能级,这使得他们具有很强的电子亲和力。此外,三维球形结构改善了富勒烯受体和聚合物给体的主链之间的接触面,这不但有利于各向异性电子传输,而且也有助于形成更好的给体/受体形貌[9]。在有机太阳电池发展的早期,富勒烯及其衍生物被广泛用作本体异质结的有机太阳电池的电子受体材料,然而在可见光区域其表现出比较弱的吸收强度,而且很难通过化学的手段来修饰改善其吸收特性,因此在活性层中主要由聚合物给体材料负责捕获光子,但效果不太显著。考虑到这一点,为了追求更高的效率,研究人员则专注于设计窄带隙聚合物给体材料(如PTB7-Th[10]和PffBT4T-2OD[11]),结果发现器件效率得到提升,基于PTB7-Th:PC71BM 和PffBT4T-2OD:PC71BM 的光伏器件效率达到了10%以上。尽管如此,当D-A共聚物通过增强D 和A 单元之间的电荷转移而减小带隙时,给体材料的HOMO 能级也会同时升高,最终导致有机太阳电池的开路电压(VOC)下降,这是因为有机太阳电池的开路电压依赖于受体的LUMO 能级与给体的HOMO 能级之差,并且呈正相关。简而言之,基于富勒烯的有机太阳电池开路电压与短路电流密度(JSC)之间难以妥协,这一问题限制了其器件效率的进一步提高。
在太阳的辐射光谱中,虽然光谱延伸至红外区,但实际上绝大多数的太阳光是在可见光区域和近红外区域内。例如,对应于1 100 nm 的吸收起始点,带隙为1.1 eV 的活性层的材料可以从太阳辐射中吸收大约77%的光子。因此,为了克服富勒烯受体这样的困境,需研究出一种有着更强的吸收强度及吸收能够红移至近红外区域的新材料,即非富勒烯受体(NIR nonfullerene acceptors,NFAs)。起初在这方面的研究中,苝二酰亚胺(PDI)由于有着较强的吸收强度、较高的电子迁移率、良好的热稳定性和可修饰的分子结构,被认为是十分有潜力的非富勒烯受体的结构单元。通常来说,PDI 类型的非富勒烯受体需要扭曲的分子构型或长烷基链的取代基,以减弱苝二亚胺较强的自聚集性,需要在良好形貌与较高的电子迁移率之间找到一个平衡点,通过改变不同类型的桥键、连接位点和PDI 的不同含量,优化了PDI 类型的非富勒烯受体的能级、带隙和聚集性,最终基于PDI 单元的受体材料获得了11%以上的器件效率[12]。但是,由于扭曲的分子结构和分子内的D-A 相互作用较弱,导致PDI 类型的非富勒烯受体的吸收范围主要在可见光区域内,带隙约为2.0 eV,限制了PDI 类型的非富勒烯受体的进一步发展。在2015 年,Zhan 等[13]以引达省并二噻吩(IDTT)为中间核、3-(二氰基亚甲基)茚-1-酮(INCN)为末端吸电基团合成了ITIC,ITIC 是一种受体-给体-受体类型的非富勒烯受体(A-D-A 型),其结合了电子受体所需要的几个特性在一个材料中,如具有较宽的吸收光谱、吸收区域延长至近红外区域,以及具有较高的吸收系数、合适的HOMO 和LUMO 能级、良好的结晶性能和较高的电子迁移率、丰富的修饰位点,使其成为了明星材料,为进一步设计和开发近红外区的非富勒烯受体做出贡献。
本文主要总结了近红外非富勒烯受体材料在有机太阳电池中的研究进展,探讨了近红外非富勒烯受体的分子设计策略、结构与性能的构效关系,以及近红外非富勒烯受体材料在三元和叠层有机太阳电池制备中的应用潜力。此外,基于当前非富勒烯受体的发展状况,对近红外区的非富勒烯受体未来的发展进行了展望。
1 近红外非富勒烯受体的中间核
为了扩展材料在近红外区的吸收,必须对非富勒烯受体的中间核进行设计和优化。设计中间核的关键在于以下几点:(1)中间核的结构要选择可供裁剪修饰的化学结构;(2)明确中间核给电子能力与结构的关系;(3)中间核的结构起着至关重要的作用,决定了非富勒烯受体的整体性能,需要注意的是中间核给电子能力的增强会使非富勒烯受体的HOMO 和LUMO 能级升高,这可能会导致给体和受体的能级不匹配,因此要注意分子结构末端基团部分的设计,因为端基INCN 可能含有卤族元素,他可以降低HOMO 和LUMO 能级而使之相匹配。本节将以中间核为核心,分别叙述ITIC 体系和Y6 体系的中间核发展进程。
1.1 ITIC 及其衍生物
1.1.1 ITIC 中间核结构
ITIC(见图3)是由一个富电子的中间核IDTT和两个缺电子的末端吸电子基团INCN 组成,其结构为一种A-D-A 分子结构,该分子结构让IDTT 与INCN 之间有着强烈的推拉电子效应,从而形成了较强的分子内电荷转移(ICT),这不仅使得ITIC 有比较窄的光学带隙(1.59 eV),还能保持合适的电子能级,其HOMO 能级为-5.48 eV、LOMO 能级为-3.83 eV。此外,IDTT 核共轭主链具有刚性和平面性,S—O 原子之间的构象锁使得ITIC 的结构更加的平面化,这有利于分子间的π-π 堆积,从而使得ITIC 具有较高的电子迁移率。ITIC 的另外一个重要的设计是在IDTT 单元上引入了4 个4-己基苯侧链,这不仅使分子具有更好的溶解度,而且还抑制了分子的过度聚集。ITIC 表现出与PC71BM 相当的光伏性能,ITIC 与PTB7-Th 共混的器件效率达到了6.8%[13]。2016 年,Hou 等[14]用PBDB-T 作为给体来替代PTB7-Th,与ITIC 共混作为活性层,由于PBDB-T 能级更加匹配的,其器件效率大幅度、、提升至11.2%,表明ITIC 类型的近红外非富勒烯受体的有着极大潜力。

图3 ITIC 的结构式和吸收光谱Figure 3 Molecular configuration and the absorption spectra of ITIC
为了使受体材料吸收更为红移,进一步减少ITIC 的带隙是突破有机太阳电池的器件效率的关键。在分子设计的层面上,需进一步增大末端基团的吸电子能力和中间核的给电子能力,更强的电子推拉效应将有利于吸收光谱的进一步红移。同时,还需综合考虑分子能级的变化、目标非富勒烯受体的结晶度和活性层共混形貌的变化。通过引入更多富电子的单元来延长π 共轭的长度,可以较好地提高中间核的醌式特性,从而提高了中间核的给电子能力。Dai 等[15]构建了有着4 个不同的中间核的稠环电子受体(F9IC)及F11IC,其稠环由5 个增至11个,研究发现:在核上加入更多的噻吩单元,可以提高稠环电子受体的吸光度和电子迁移率,同时也会使稠环电子受体的HOMO 能级提高,溶解度会逐渐变差;此外,随着中间核上的稠环的数量增加,会逐渐到达一个饱和点,而延长π 共轭长度不会使得中间核的给电子能力增加,如F9IC 的吸收光谱则几乎与F11IC 的吸收光谱重叠。所以,中间核的共轭长度十分重要,设计近红外非富勒烯受体时需要注意。
1.1.2 ITIC 中间核的异构化
异构化可以通过改变中间核的原子/官能团的空间顺序或拓扑结构来调整分子的电子结构和轨道分布,从而改变中间核的给电子能力。将IDTT 的核异构化成苯并(二环戊二烯)单元,从而构建出ITIC 的异构体,即BDT-IC 或者NFBDT(见图4),通过异构化方法使BDT-IC 表现出更强的分子内电荷转移和带隙减小为1.53 eV(ITIC 的为1.59 eV),之所以具有强的分子间和分子内电荷转移效应是因为分子间的堆积更紧密,具有紧凑的晶体结构,与ITIC 相比,基于BDT-IC 的有机太阳电池的电子迁移率提高了5 倍,器件效率超过10%、电流密度JSC显著增加[16]。Wang 等[17]用异构化的方法研究近红外吸收的非富勒烯受体,通过异构化将苯并[1,2-b:4,5-b']二噻吩[3,2-b]并噻吩和苯并[1,2-b:4,5-b'']二噻吩重新设计为FNIC1 和FNIC2,研究发现:环戊二烯单元在核中的位置变化对分子性质起着重要的作用,FNIC2 的吸收边发生很大的位移,而且带隙也减少了0.1 eV;此外,FNIC2 具有较高的结晶度,并能与PTB7-Th 共结晶,这不仅增强了载流子迁移率,而且有助于在本体异质结薄膜中形成合适的/受体垂直分布,从而使在没有任何后处理的情况下的器件效率超过13%。Lin 等[18]报道了两种基于硒酚官能化的IDTT 核的非富勒烯受体异构体,分别是SRID-4F 和TRID-4F(见图4),其都表现出了与IT-4F 相似的吸收红移,硒(Se)原子位于核外侧的SRID-4F 比硒原子位于核中部的TRID-4F 具有更宽的吸收,因此与基于PBDB-T-2F:TRID-4F 的OSCs 相比,基于PBDB-T-2F:SRID-4F 的OSCs 获得较高的JSC。

图4 ITIC 衍生物的结构式Figure 4 Molecular structures of ITIC derivatives
1.1.3 增强ITIC 中间核给电子能力
增强中间核给电子能力最有效方法是在核中加入富电子单元。因为芳香化合物具有芳香性,在IDTT 的核中含有苯的部分则有很高的稳定性,但其给电子能力很低,如果可以使用其他富电子单元替换掉核中含有苯的部分(如噻吩及其衍生物),那么核的给电子能力会有显著提升。6TIC-4F(见图4)就是对IDTT 的核进行修饰,用噻吩并[3,2-b]噻吩单元来取代核中间的苯而合成,研究表明6TIC-4F具有很窄的带隙(1.24 eV),并且其在近红外区有着较强的吸收能力(吸收边超过1 000 nm),这使得JSC达到了23 mA·cm-2,并且VOC的能量损失很小只有约0.517 eV[19]。Xiao 等[20]通过把醚桥加入到核中,发现了另一种高性能的近红外区非富勒烯受体COi8DFIC(见图4),当与PTB7-TH 共混,在吸收光谱超过1 000 nm 时EQE 仍有较好的响应,对应的JSC高达26.12 mA·cm-2。此外,通过把富电子单元加入核中的非富勒烯受体还有很多,如IEICO-4F[21]、DTPC-DFIC[22]、SN6IC-4F[23]和其他材料等[24-28]。
在近红外区非富勒烯受体的研究中发现,减少非富勒烯受体的带隙会降低LUMO 的能级,使得最终器件的VOC降低,然而这样的问题也出现在用于富勒烯体系的近红外聚合物给体中。因此,设计近红外非富勒烯受体时,要同时实现较高的JSC和VOC,同时要有较低的能量损耗Eloss(Eloss是指光学带隙与元电荷VOC之间的差值),这是有机太阳电池的PCE进一步突破的关键。Sun 等[29]首次将了二噻吩并[3,2-b:2',3'-d]吡咯单元取代了IDTT 核中噻吩并[3,2-b]噻吩部分,合成了近红外非富勒烯受体INPIC-4F(见图4),由于吡咯的给电子能力比噻吩的要强,与不含有吡咯的INIC3 相比,INPIC-4F 有更加窄的带隙(INIC3 的带隙为1.46 eV、INPIC-4F的带隙为1.39 eV),基于PBDB-T:INPIC-4F 的光伏器件效率超过13%,有着平衡且较高JSC和VOC,其Eloss相对于其他基于PBDB-T 的OSCs 比较低(Eloss为0.54 eV)。为了进一步研究吡咯在减少Eloss的作用,Gao 等[30]设计了一种仅含有单个吡咯的非富勒烯受体N7IT。在与a-IT 进行全面的比较后发现,引入吡咯单元可以较好地降低Eloss(从0.718 eV降低至0.573 eV),显著抑制无辐射复合损耗(从0.370 eV 降至0.266 eV)[31]。通过对N-烷基链的优化和引入氟化的端基,得到了N7IT 的衍生物IDTP-4F,其器件效率达到15%。在吡咯中的氮原子(N)不仅可以提高核的电子云密度,而且可以引入侧链增加溶解性。Ma 等[32]利用这一优势,用吡咯单元取代普通稠环中间核中的环戊二烯,设计合成了一系列高性能的近红外非富勒烯受体(命名为M 系列),与基于IDTT 型中间核的非富勒烯受体相比,M 系列具有更好的分子结晶度和更短的π-π 堆积距离,这是因为N-烷基链比sp3杂化的碳烷基链具有较小的空间位阻,从而保证了分子间的有效堆积,其中M34 有着较小的带隙(1.39 eV)和较低的HOMO能级(-5.60 eV),这使得其与PM6(PBDB-T-2F)能够很好地匹配,基于PM6:M34 的光伏器件效率能达到15.24%,以及VOC为0.91 eV、JSC为23.63 mA cm-2、Eloss也低至0.48 eV。通过对烷基链部分的设计,进一步优化了BHJ 的形貌,基于PM6:M3 的器件FF 值有很大的提升,且器件效率达到16.66%[33]。此外,还有一种关于不对称分子设计方法,虽然不能直接增加核的给电子能力,但可以更精确地控制非富勒烯受体的结构,从而微调其性质,比如影响BHJ形貌的分子结晶度[34]。Gao 等[35]曾通过将单个噻吩单元加入IDTT 核中或将单个噻吩单元从核中去除,从而合成了两个不对称的中间核(IDT6CN-M 和IDT8CN-M),由于分子中的正负电荷中心不重合,从而导致产生的偶极矩较大。然而,偶极矩对活性层的形貌是有影响的,主要体现在以下方面:(1)偶极和偶极之间的相互作用,会增强分子之间的π-π 堆积能力;(2)偶极矩的方向几乎垂直于分子的长轴,诱导分子形成更有利的堆积方式,从而使得分子间的π-π 堆积更有序,这样的偶极矩的协同效应优化了活性层的形貌,使得其具有较高且平衡的载流子迁移率,从而增加OSCs 的FF 值。(3)较大的偶极矩也会增加材料的介电常数,从而降低了激子结合能,使有机太阳电池的解离效率得到提升,并且同时降低了能量损失。若要设计更高性能的近红外非富勒烯受体,需要结合上述所说的几种方法对中间核进行设计,例如:延长π 共轭长度,在能级匹配的情况下增加给电子能力;将中间核异构化,从而可以改变中间核的给电子能力;将富电子的单元引入中间核,以及加入能减少带隙且要有较低Eloss的单元;将中间核设计成不对称,从而调控其形貌等。ITIC 及其衍生物的光学带隙、能级和光伏参数列于表1。

表1 ITIC 及其衍生物的光学带隙、能级和光伏参数Table 1 Optical bandgap,energy levels,and photovoltaic parameters for ITIC and its derivatives NIR NFA
1.2 Y6 及其衍生物的中间核
2019 年,Zou 等[36]报道了命名为Y6 的一种ADA'D-A 型的NFA,首次将缺电子的苯并噻二唑(BT)单元加入富电子的中间核中(见图5),Y6 表现出较好的性能,有着1.33eV 的窄带隙和比较合适的HOMO、LUMO 能级(分别为-5.65 eV 和-4.10 eV),在纯膜和共混薄膜中都有着较高的电子迁移率和合适的结晶度,单结有机太阳电池器件效率达到15.7 %,其中JSC 高达25.3 mA·cm-2,Eloss 仅为0.49eV。
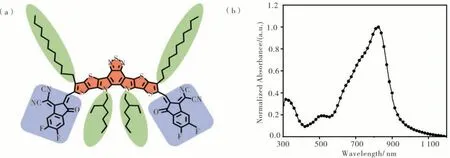
图5 Y6 的结构式和吸收光谱Figure 5 Molecular configuration and the absorption spectra of Y6
Y6 受体的出现使得全小分子体系和给体/小分子受体体系迅速发展,推动有机太阳电池的发展。由于Y6 有着优异的光伏性能,在分子设计上有着几个重要的关键点:(1)两个吡咯单元使中间核具有很强的给电子能力和良好的溶解性,并且这可能减少能量损失,获得较低的Eloss;(2)空间位阻效应是由两个N-烷基链太近引起的,导致中间核的结构略微扭曲,这在一定程度上可以避免分子过度聚集;(3)BT 单元的引入不仅使Y6 有着相当低的HOMO能级,以此使得空穴能有效地传输,而且还通过S—N 非共价的相互作用增强了分子间的相互作用;(4)噻吩并[3,2-b]噻吩中的侧烷基链抑制分子构象的变化,提高了中间核和端基之间乙烯基键的稳定性;(5)Y6 的半月型的分子结构有利于其自身的有序堆叠。在对Y6 单晶的研究中发现,Y6 可以形成连续的、规则的三维结构,分离的电子和空穴传输的通道分别分布在DA′D中间核和端基中。此外,由于端基和中间核之间有明显的重叠,Y6 分子表现出较强的分子间相互作用,这与传统的基于IDTT 的NFAs 有很大不同,增强了相邻分子之间的电子耦合,从而促进了电荷传输。最重要的是,通过旋涂制备的Y6,其三维网络结构可以保存在纯膜和共混膜中[37]。
中间核中的缺电子单元,对Y6 型NFAs 的性能起着至关重要的作用。比较常见的缺电子单元,如苯并[d][1,2,3]噻三唑(BTz)、喹喔啉(QX)、苯并[c][1,2,5]硒二唑(BS)、苯并[c][1,2,5]恶二唑(BO)和邻苯二甲酰亚胺(IID)。通过将Y6 的BT单元替换成BTz 单元,合成了非富勒烯受体Y11(见图6),由于BTz 单元的吸电子能力比BT 单元弱,Y11 的带隙可以进一步减小到1.31 eV、吸收边超过950 nm,Y11 与PM6 共混时器件效率为16.54%、VOC为0.832 V、JSC为26.74 mA · cm-2、FF 为74.33%,表明Y11 在抑制非辐射复合和能量无序性方面有较好的效果[38]。然而,最新的研究表明[39-40]:BTz 单元上较大的N -烷基链对基于BTz 的NFAs的堆叠有着较大的影响,导致FF 因子不会太高;此外,Y6 中间核上的噻二唑单元可以通过化学还原法转化为二胺中间体,再将二胺中间体分别与乙二醛和二氧化硒反应,可以很容易地得到含有喹喔啉单元或苯并[c][1,2,5]硒二唑单元的Y6 型非富勒烯受体。Zhou 等[41]报道了核中含有缺电子喹喔啉单元的AQx-2(见图6),AQx-2 的带隙略有增加为1.35 eV,这导致JSC有小幅降低、VOC小幅提升,AQx-2 的纯膜和共混膜中有着紧凑的π-π 堆积,这促进了高效且平衡的电荷传输,从而使得AQx-2 的器件有较高的FF 值。Chen 等[42]设计并合成了两个具有新型的非富勒烯受体(CH4 和CH6),二者均具有新型扩展中心核单元且中心单元上具有氟化作用,采用苯并噻二唑的高效原位吩嗪转化可以方便地构建CH4 和CH6 中新出现的吩嗪中心单元,与CH4 相比,中心单元氟化的CH6 表现出能级下移、更强的分子π-π 相互作用、结晶度增加,表现出优异的纤维网络形态,基于PM6:CH6 的二元器件效率达到了18.33%、VOC为0.875 V、JSC为26.62 mA·cm-2、FF 值为78.4%,基于PM6:CH6 的光伏器件效率为16.42%、VOC为0.888 V、JSC为26.11 mA·cm-2、FF相对较低,仅为71.1%。Zhang 等[43]设计合成了中间核含有苯并[c][1,2,5]硒二唑单元的Y6Se,相对于Y6 和Y6Se 有更加窄的带隙(1.31 eV)和更高的电子迁移率,这归功于Se 和Se—Se 键的相互作用,使得给电子能力比S 原子的要强,此外Y6Se 有着更陡的起始吸收波长及较低的乌尔巴赫能量(Urbach energy,20.4 meV),表明能量无序性程度更小,在没有任何后处理的情况下基于D18:Y6Se 的器件效率达到了17.7%、JSC接近28 mA·cm-2。此外,有研究对中间核的噻吩并[3,2-b]噻吩单元(TT)进行改造,将噻吩并[3,2-b]噻吩单元与吡咯的融合,部分地促进了Y6 的给电子能力和中间核的堆积。CH1007(见图6)是一种高性能的Y6 衍生物,其中间核的两侧的噻吩被硒酚取代,与Y6 相比,CH1007有更强的面面堆积相互作用和更短的π-π 堆积距离,基于PM6:C6H1007:PC71BM 的器件效率达到了17.07%、JSC高达27.48 mA·cm-2,以及比基于PM6:Y6:PC71BM 的光伏器件更低的Eloss[44]。然而,在含呋喃的Y6 中间核中,取代侧边噻吩会显著降低光伏性能,因为呋喃的氧原子与端基的羰基之间缺乏非共价的相互作用,CH1007 的异构物Y6-2SE 是通过将Y6 中间核中部的噻吩单元替换为硒酚合成的,其吸收光谱和CH1007 一样会吸收蓝移,而这不利于增大JSC[45]。此外,不对称地改变π 核共轭,使用噻吩单元、二噻吩并[3,2-b:2',3'-d]噻吩单元和并四噻吩单元来替换Y6 衍生物中间核的噻吩并[3,2-b]噻吩单元,其中Y6 衍生物有着不同大小的核和构象,如AY6、BP5T-4F、ABP4T-4F、BP6T-4F 和ABP6T-4F(见图6)[46-48]。

图6 Y6 衍生物的结构式Figure 6 Molecular structures of Y6 derivatives
目前,Y 系列小分子受体的有机太阳电池的能量转换效率已有了较大的突破,但是该类型的材料在受热或者光照条件下活性层的形貌会变得不可控,从而使得器件的性能和稳定性收到了较大的影响。活性层微观形貌不稳定的问题,国内外许多研究者采用将小分子受体聚合物化的策略,所得聚合物受体不仅能够维持小分子受体的效率,也表现出更好的光稳定性或热稳定性。Liang 等[49]出了非富勒烯受体的低聚策略,合成了一系列具有确定化学结构的Y 系列齐聚物,实现了对材料热力学性能、结晶特性和分子堆积的有效调控。在老化实验中,OY3 能够保持优秀的热稳定性和光照稳定性,基于OY3 的有机太阳电池在光照下工作1 000 h 后仍能保持超过90%的初始效率,这得益于OY3 具有更高的热转变温度和更有序的微观形貌,该结果是在本领域中报道的第一个兼具高效率(大于15%)和高使用寿命(大于15 年)的有机太阳电池。Y6 及其衍生物的光学带隙、能级和光伏参数列于表2

表2 Y6 及其衍生物的光学带隙、能级和光伏参数Table 2 Optical bandgap, energy levels, and photovoltaic parameters for Y6 and its derivatives NIR NFA
2 近红外非富勒烯受体的端基和侧链
2.1 端基的优化和设计
氟原子(F)不仅有着很强的电负性,还有相对较小的原子大小。因此,在不造成严重空间位阻的情况下,氟原子在INCN 上的取代可以改善分子的吸电能力。此外,末端基团的氟化是通过诱导非共价键相互作用,从而加强分子间的堆积,如F—H、F—S 和F—O 的相互作用[50],并且促进极化以减少电子空穴对之间的库仑力,使得激子的解离更加的高效[51]。
IT-4F(见图7)是通过氟原子取代ITIC 的端基5,6 位点所合成的。与ITIC 相比,IT-4F 的带隙减小(1.55 eV)、吸收系数增强、π-π 堆积更加有序紧密,当IT-4F 与氟化的给体PBDB-T-SF 相配对时对应的器件效率达到了13%、JSC超过20 mA·cm-2[52]。当逐渐提高端基的氟取代基的数量时,零个氟原子的ITIC 到六个氟原子的ITIC-6F,随着吸收系数增加到2.0×105M-1·cm-1,非富勒烯的受体的带隙可以减少到1.51 eV,但只是略微地降低了LUMO 能级,同时JSC和VOC得到提高。更重要的是,氟原子取代数量的增加使得分子间的堆积更紧密,从而使π-π 堆积的d-spacing 距离更短,这不仅促进了分子间的电子耦合,使分子间的电子传递更高效,而且显著地降低了重组能[53]。
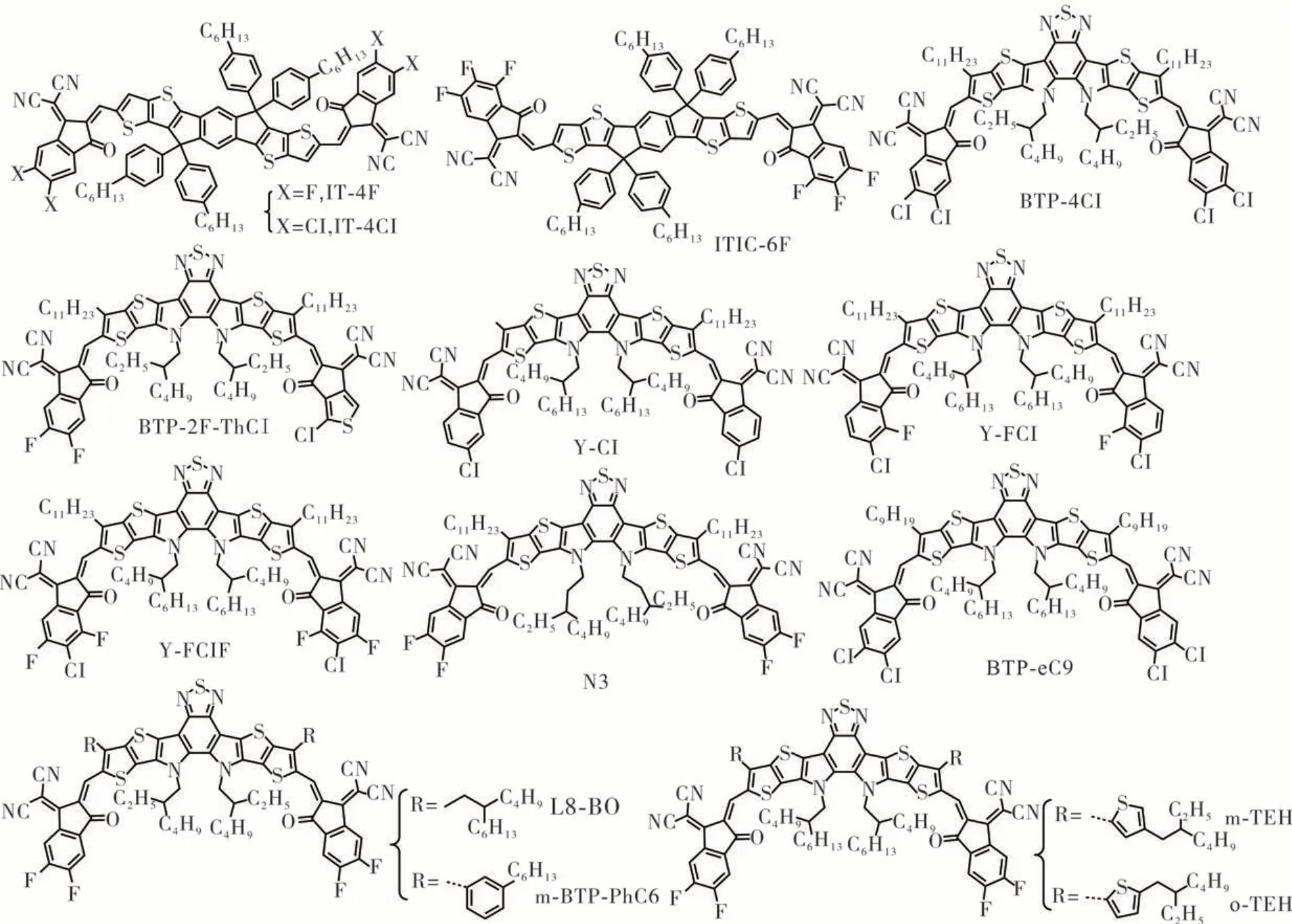
图7 ITIC 衍生物和Y6 衍生物的结构式Figure 7 Molecular structures of ITIC derivatives and Y6 derivatives
氯(Cl)是与氟同属一个主族的元素,有着稍弱的电负性和更大一些的原子半径。然而,由于C—Cl键的极性更强,氯取代的端基的偶极矩比氟取代的偶极矩大(2.77 D vs 2.26 D),这使得IDTT 中间核到氯化的端基的ICT 得到了增强,与IT-4F 相比,IT-4Cl 的吸收光谱(见图7)被拓宽至了40 nm 和有着1.48 eV 的窄带隙。此外,氯取代基的偶极矩越大,π-π 堆积的会更加地有序、电子迁移率更高,基于PBDB-T-2F:IT-4Cl 的器件有着更加高的外量子效率及更好的激子解离和电荷传输特性,电流密度JSC超过了22 mA·cm-2[54]。
Y6 型非富勒烯受体的吸电子端基有着十分重要的作用,不仅可以改变材料的带隙和能级,还在调控分子结晶性质方面也发挥着重要作用,并对器件的PCE 有着很大影响。Cui 等[55]报道了一种高性能的Y6 型受体BTP-4Cl,使用Cl 原子来替换端基上的F 原子所合成的,BTP-4Cl 在吸收和π-π 堆积方面比Y6 更有优势,实现了器件中JSC和VOC的同时增强,显著地抑制非辐射能量损失(VTP-4Cl 的为0.205 eV、Y6 的为0.230 eV)。通过不对称地将Y6一侧的端基替换为含有噻吩的端基,Luo 等[56]报道了不对称的Y6 衍生物BTP-2F-ThCl(见图7),其有着平衡的JSC和VOC,并微调了共混膜的形貌,器件效率超过17%。Yan 等[57]采用逐步氯化/氟化策略成功合成了两个具有良好确定的氟化和氯化位置的新型杂卤化端基(分别命名为o-FCl-IC 和FClFIC),随后又分别合成了3 种基于Y 系列中心核的无区域异构体的小分子受(Y-Cl、Y-FCl 和Y-FClF),其具有3个不同的杂卤化端基,随着杂卤化端基中氟取代数目的连续增加(从Y-Cl 到Y-FCl 和Y-FClF),分子吸收光谱逐渐红移,HOMO 能级和LUMO 能级降低,同时逐渐提高了纯膜中的电子迁移率,PM6:YFClF 共混膜在分子堆积上表现出最有序和最优先的取向,在合适的相分离下表现出最强的结晶倾向,在3 种共混膜中具有最高和最平衡的电荷迁移率,基于PM6:Y-FClF 的器件效率达到了17.65%,明显高于基于PM6:Y-FCl(16.00%)和PM6:Y-Cl(14.47%)的器件效率。通过优化端基可提高Y6 衍生物的光伏性能,如BTP-ClBr[58]、BT-BO-L4F[59]、BTIC-2Cl-γCF3[60]。
2.2 侧链的优化和设计
在Y6 中间核侧边的噻吩并[3,2-b]噻吩单元上的侧链和在N 的烷基链会影响分子的溶解性和结晶性,通过优化侧链来调控非富勒烯受体与聚合物给体的共混性是一种获得最优BHJ 形貌的简单而有效的方法,从而使器件获得更高的FF 值。Jiang等[61]对Y6 中间核侧链进行了改造设计,优化了Y6的分子结构,研究了烷基链的支链点对分子性质和器件性能的影响,表明具有3 个分支点的烷基链的非富勒烯受体(N3)具有更好的溶解性和共混膜形貌,基于N3 的器件在其二元和三元器件中均实现了超过16.42%的效率。Cui 等[62]进一步报道了一套烷基链优化,延长N-烷基从2-乙基己基(EH)到2-丁基辛基(BO),并将噻吩并[3,2-b]噻吩单元上的侧十一烷基缩短为壬基,得到BTP-eC9(见图7),其器件实现了17.8% 的效率及非常高的FF 值(81.1%)。相对于噻吩并[3,2-b]噻吩单元上的直链烷基链,支链和芳香侧链会造成更大的空间位阻,并将影响其溶解性和堆积。Li 等[63]对Y6 进行了其他方面的优化,用具有不同支链的烷基链来改变噻吩并[3,2-b]噻吩单元上的呈线性的烷基链,设计合成了一种高性能的Y6 衍生物L8-BO(见图7),L8-BO 具有更紧凑的堆积结构,基于PM6:L8-BO 的光伏器件效率超过18.32%、VOC为0.87 V、JSC为25.72 mA·cm-2、FF 值为81.5%。Chai 等[64]用十六烷基苯基取代TT 单元上的烷基链,设计合成了另一种Y6 衍生物m-BTP-PhC6,通过调控己基在苯环上的相对位置(邻位、间位和对位),研究了侧链的位置对分子平面性、吸附特性和分子间堆积的影响,基于m-BTP-PhC6 与有宽带隙聚合物PTQ10的器件效率实现了17.7%、VOC为0.883 V、JSC为25.3 mA·cm-2、FF 值为79.3%。Kong 等[65]通过在α-或β-位置引入带有2-乙基己基取代的噻吩共轭外侧链,设计并合成了2 种新型A-DA'D-A 型异构体消小分子受体(分别命名为o-TEH 和m-TEH),当以基于喹啉结构的宽带隙聚合物PBQ6 作为给体时,基于m-TEH的OSCs 器件比基于o-TEH 的器件具有更合适的相分离和增强的分子堆积,获得了更高且更平衡的空穴和电子迁移率及更长的载流子寿命和载流子复合,基于PBQ6:m-TEH 的二元器件效率达到了18.51%、VOC为0.880 V、JSC为26.61 mA·cm-2、FF为79.03%。ITIC 衍生物与Y6 衍生物的光学带隙、能级和光伏参数列于表3。

表3 ITIC 衍生物与Y6 衍生物的光学带隙、能级和光伏参数Table 1 Optical bandgap, energy levels, and photovoltaic parameters for ITIC derivatives and Y6 derivatives NIR NFA
3 近红外非富勒烯受体的三元和叠层有机太阳电池
从电流-电压取舍的角度来看,制备一种只有一个给体和一个受体的二元活性层材料,要同时拥有较高的VOC和JSC是非常困难的。因此,为了平衡VOC和JSC,可以在二元共混物中添加一种具有其他优势的且与其兼容的第三组分,这不仅可以有优化光学吸收,还有助于调整活性层的BHJ 形貌,从而实现同时提升VOC、JSC、FF 和PCE。虽然三元策略可以通过使用带隙不同的材料的组合有效地提高器件的效率,但在早期的研究中缺乏像ITIC 和Y6 这样高效的近红外吸收的受体,三元有机共混太阳电池的效率提升仍然受到较大限制。ITIC、Y6 及其衍生物的出现为三元有机共混策略提供了很多思路,可以充分利用不同材料的优势来制备。例如,PM6 与Y6 共混膜在300—500 和600—800 nm 范围内的吸收强度相对较弱,导致在这些区域的EQE 响应较低,而Y6 的LUMO 能级较低也使得VOC相对较低。考虑到这些因素,可以在PM6:Y6 体系中掺入带隙更宽的兼容性好的受体材料,既弥补了为提升JSC而活性层的吸收不足,而且还有助于提高VOC。Yu 等[66]将PC61BM 添加到PM6:Y6 共混膜中,制备了一种三元有机共混太阳电池,与基于PM6:Y6 的二元器件相比,其光伏参数全部增强且有16.5%的效率。MF1 受体在600—800 nm 范围内表现出较为合适的带隙,并具有较强的吸收强度[67]。An 等[68]报道了另一种三元有机共混太阳电池,由于MF1 和Y6 之间形成了类合金的结构,三元有机共混太阳电池器件最终实现了17.2% 的效率。在最新的研究[69]中,通过采用不同的第三组分和优化的Y6 衍生物,基于PBQx-TF:eC9-2Cl 体系的三元有机共混太阳电池器件已经实现了超过19%的效率。此外,Sun 等[70]通过在基于PM1:L8-BO 主体混合物中引入一种新型不对称非富勒烯受体BTP-2F2Cl,从而制备出高效的三元有机太阳电池,同时还系统地研究了共混物的相分离形貌,并对光活性层进行了物理动力学测量并发现:性能最好的三元器件效率达到了创纪录的19.17%(认证效率18.7%),而JSC为27.15 mA·cm-2、FF 为80.14%、VOC为0.881 V;同时,三元器件的效率明显高于二元器件的效率,基于PM1:L8-BO 与PM1:BTP-2F2Cl 的器件PCE 分别为18.51%和18.40%,以及三元体系表现出更好的运行稳定性,在250 h 的光照条件下效率仍能保持初始值的83%。
此外,叠层有机太阳电池将2 个或者2 个以上的单结器件通过串联或者并联的方式叠加在一起,是另一种提升器件效率的有效方法。叠层有机太阳电池可以克服单结电池的厚度限制,通过叠加吸收互补的前后电池实现效果更好的太阳能捕获。由于缺少高效的近红外非富勒烯受体,叠层有机太阳电池的发展受到了极大的限制,其器件效率为11.6%,与单结器件效率的记录相同[71]。Meng等[72]报道了一种叠层有机太阳电池,通过把一种带隙非常窄的受体材料COi8DFIC 加入到后电池,这使得其器件效率提高到了17.3%。Liu 等[73]研究了一种有效的互连层,降低后电池的最优厚度,显著地抑制了电荷复合,基于COi8DFIC 的叠层有机太阳电池器件效率实现了18.7%,并且FF 达到了较高的78%。近年来,通过调节光强分布和结合兼容不同的受体材料,在前电池实现较高的VOC和在后电池实现较高的JSC,最后叠层有机太阳电池器件效率实现了19.6%的[5]。
4 总结与展望
具有近红外吸收的非富勒烯受体,在提高有机太阳电池器件效率上起着十分重要的作用。这种类型的材料具有A-D-A 分子构型,由吸电子的端基、富电子的中间核和可溶性侧链构成。首先,卤化端基与具有较强的给电子能力的核的结合,显著提高了A 和D 单元之间的分子内电荷传输强度及具有更宽的近红外吸收,使有机太阳电池有更高的JSC。其次,在对中间核的设计方面,含有吡咯单元的中间核可以较好地平衡VOC和JSC,从而有效地降低Eloss。目前,Y6 及其含有BT 中间核的衍生物是目前较为先进的近红外非富勒烯受体,Y6 具有合理的分子设计及独特的三维堆积架构,所以有着优异的光伏性能,其不仅使二元有机太阳电池实现了超过18%的效率,还有三元和叠层有机太阳电池器件实现了超过19%的效率。在此,为了实现更高的器件性能和促进有机太阳电池的实际应用,应该进一步设计或优化近红外吸收的非富勒烯受体。
目前,近红外非富勒烯受体的化学不稳定性源于中间核和端基之间活跃的双键,其容易被亲核物质攻击,在光照条件下可能发生环氧化,然后降解为非活性成分[74]。这内在的缺陷将严重影响光伏器件的稳定性和寿命,限制了未来的商业应用。从材料设计的角度来看,近红外非富勒烯受体的这一短板是可以克服的。例如:可通过用惰性基团取代酸性的氢原子,降低双键的反应(如烷基、烷氧基、芳香族或氰基);与中间核融合双键,进一步固化分子;研发新型的吸电子端基,用单键来连接中间核和端基[75]。
为了实现有机太阳电池的商业化,必须考虑光功能材料合成成本的问题。一般来说,复杂的分子结构意味着较高的生产成本。因此,通过选择性价比高且容易获得的原料,简化NIR NFAs 的结构,则可以大大地降低生产成本。最近,Hou 等[76]报道了一种基于四噻吩单元的近红外非富勒烯受体,其器件效率超过15%,这不仅证明了这种寡聚噻吩作为中间核的有效性,同时也为基于不含噻吩寡聚物的非富勒烯受体的设计提供了方向,表明以寡聚噻吩作为中间核的近红外非富勒烯受体的发展具有广阔的前景。
由于Y6 具有良好的光伏性能,很大程度上依赖于其规整的三维堆叠框架。如果分子结构进行设计的时候破坏了其堆叠模式,这很有可能会降低器件的性能,使Y6 在分子结构设计这方面受到了限制,但是可以围绕分子堆叠这方面进行研究来提升器件的性能。为了进一步发展NIR NFAs 以获得更高效率,需要研发更多吸收红移的材料。最近有报道[77]指出,在不牺牲电荷产生效率的情况下,像Y6 这样高性能的NFAs 可以获得相当低的非辐射VOC(约为0.2 eV)损失,所以当进一步减小材料的带隙时需要保留这种特性,以避免较大的非辐射VOC的损失[78]。另一方面,由于有机材料的吸收范围相对较窄,其EQE 曲线通常呈“M”形,而非无机材料呈现阶梯形吸收曲线。一个合适的第三组分通常有着稍大一些的带隙[79-80],或是具有覆盖范围较广的吸收光谱的二维共轭材料,对于进一步提升有机太阳电池的PCE 也是十分必要的。Y6 即使没有给体材料的帮助下,也能够产生自由载流子。然而,在单组分的Y6 型有机太阳电池中,活性层材料的电荷提取率远低于辐射复合速率。因此,应该研究有助于Y6 电荷提取的界面层材料,研发新的化学结构来进一步降低有机半导体固有的较大的激子束缚能,这些都是未来材料的设计方向。
