口服大黄酸超分子纳米粒的制备与表征及其巨噬细胞靶向性能研究
2024-01-03聂文彪林大胜高飞
聂文彪,林大胜,2,高飞
大黄酸(Rhein,RH),又称大黄苷,是一种天然植物蒽醌,主要存在于蓼科植物中,为大黄、何首乌、芦荟和番泻叶等多种中药的主要生物活性成分之一[1]。其化学结构含有羟基和羧基,极性较强,呈弱酸性,为咖啡色针状结晶,升华后为黄色针状结晶,几乎不溶于水,微溶于乙醇、苯、氯仿、乙醚、石油醚,能溶于吡啶、碳酸氢钠水溶液等碱溶液中。现代药理学研究表明,RH药理用途十分广泛,具有抗炎,抗肿瘤,抗菌,降糖调脂,调节免疫和抗动脉硬化等作用,在临床上用于淋病,炎症性疾病和代谢性疾病的辅助治疗,其可培植衍化多种药物,是治疗骨关节炎的良药“双醋瑞因”的主要原料,也是配制保健品的最佳原料,可起到降脂减肥、通便排毒、清洁内环境的效果[2-5]。由此可见,RH在医药领域和功能保健食品领域有着良好的发展前景。然而,RH溶解性差、生物利用度低,靶向性能差及大剂量服用具有肝毒性风险等缺陷,很大程度上限制了它的临床用途。
近年来,超分子聚合物因其在生物技术、纳米传感、药物传递和基因诊断和治疗等领域的潜在应用而受到了广泛的关注[6]。由聚合物自组装形成的超分子纳米通常具有规则的球形核壳结构。它们的疏水核可以与治疗药物在物理或化学上相互作用,从而可作为封装疏水性药物的纳米级容器,而亲水壳层可以与溶剂相互作用,提高纳米粒子的稳定性。超分子聚合物通常是高度取向的,并通过非共价弱相互作用结合在一起,如氢键、主客体相互作用、π-π堆积、金属配位键和静电相互作用。其中,主客体相互作用具有选择识别性,需要主体和客体分子之间大小和结构的互补[7,8]。目前,环糊精与金刚烷之间的主客体相互作用已得到广泛认可,国内外学者以此为契机,开发出了多种智能口服纳米递药系统[9-11]。基于以上背景,本研究以环糊精与金刚烷这对主客体分子为基础,分别对二者加以修饰,通过主客体相互作用自组装包载疏水性药物RH,制备RH超分子纳米粒子,并采用壳聚糖-果胶复合聚电解质(CP)对其进行包裹,以能够实现安全稳定的口服递送;最后,对制备出来的纳米粒子进行理化性质表征,同时也对该纳米粒子的巨噬细胞靶向性能进行了研究。
1 实验仪器与材料
1.1 实验仪器
Litesizer 500激光粒度分析仪,安东帕(上海)商贸有限公司;Agilent 1260型高效液相色谱仪,美国Agilent公司;ESJ205-S电子分析天平,沈阳龙腾电子有限公司;HMS-901D6联多点加热磁力搅拌器,深圳市博大精科生物科技有限公司;JEM 2100F透射电子显微镜,日本电子株式会社;TSC SP8激光共聚焦显微镜,德国Leica公司;AVANCE NEO 700M核磁共振波谱仪,D8 X射线衍射仪,德国Bruker公司;IRTracer-100傅里叶变换红外光谱仪, 岛津企业管理(中国)有限公司。
1.2 实验材料
单(6-氨基-6-去氧)-β-环糊精(C D,批号Z220820),山东滨州智源生物科技有限公司; 硫代羟基乙酸酐(TA,批号M220522),上海麦克林生化科技有限公司;维生素E聚乙二醇琥珀酸酯(TPGS,批号C220318),上海阿拉丁生化科技股份有限公司;金刚烷胺(AD,批号R220416),上海罗恩科技发展有限公司;叶酸(FA,批号XC220312),深圳金富源生物科技有限公司;大黄酸(RH,批号wkq22052602,纯度≥98%),四川维克奇生物科技有限公司;香豆素6(C6),上海谱振生物科技有限公司;1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(EDC)、N-羟基琥珀酰亚胺(NHS)、4-二甲氨基吡啶(DMAP),西格玛奥德里奇(上海)贸易有限公司;二甲基亚砜(DMSO),北京汇海科仪科技有限公司;甲醇,北京迪科马科技有限公司。
2 方法与结果
2.1 主体聚合物CD-TA-TPGS的合成与分析
根据酰化反应的基本原理,采用TA作为酰化试剂合成主体聚合物CD-TA-TPGS[12]。具体如下:将CD、TA(按2:3的比例)和少许催化剂DMAP一起溶解在20 ml DMSO中,并向其中滴加少许三乙醇胺共同搅拌24 h;然后利用透析技术除去反应溶液中的DMSO和少许杂质,再通过真空冷冻干燥得到中间体CD-TA。之后,将得到的CD-TA粉末复溶于20 ml DMSO中,并向其中加入与CD等量的TPGS和少量催化剂EDC与DMAP,在溶解完全后在50 ℃下反应24小时,然后同样经透析技术去除DMSO和杂质,再通过真空冷冻干燥最终获得主体聚合物CD-TA-TPGS。通过核磁共振波谱仪和傅里叶变换红外光谱仪验证CD-TA-TPGS的成功合成,结果见图1~2所示。图1显示了CD、CD-TA、TPGS和CDTA-TPGS四种物质的核磁共振氢谱图。在CD-TA谱图中,CD的质子峰几乎全部被包括在内,并在8.61 ppm(a)处出现了一个新的质子峰,代表新生成的酰胺基团上氢的信号峰,表明CD和TA成功接枝在一起。此外,从CD-TA-TPGS谱图中可以看出,0.83 ppm(c)是TPGS维生素E长链甲基端氢的信号峰;1.00~1.71 ppm为维生素E长链上不同环境亚甲基氢的信号峰; 2.52 ppm(d)是维生素E苯并二氢吡喃环4位亚甲基上氢的信号峰,2.66 ppm(e)和2.84 ppm(f)是TPGS琥珀酸链上两个亚甲基上的氢信号。与CD-TA和TPGS谱图相比,CD-TA-TPGS谱图在5.54~5.78 ppm(b)处同样出现了CD中的羟基质子峰,并且仲酰胺基团的氢信号峰(a)仍然存在,这些充分证实了TPGS接枝在CD-TA上,成功合成了主体CD-TA-TPGS 聚合物。傅里叶变换红外光谱图也进一步证实了我们的分析。如图2所示,与CD光谱相比,CD-TA谱图在1651 cm-1处出现新的峰(C=O),表明TA成功反应并接枝在CD上。在TPGS的谱图中,1736 cm-1被强吸收,这是酯羰基的峰值(O-C=O),1115 cm-1是醚键(C-O-C)的拉伸振动。与CD-TA谱图相比,CD-TA-TPGS 谱图具有1737 cm-1酯羰基(O-C=O)峰和1112 cm-1醚键(C-O-C)峰,这也表明TPGS通过酯化反应成功与CD-TA接枝形成CD-TA-TPGS主体聚合物。

图1 CD,CD-TA,TPGS和CD-TA-TPGS的核磁共振氢谱图
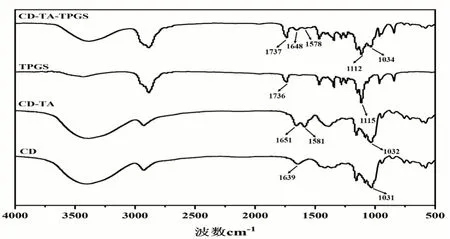
图2 CD,CD-TA,TPGS和CD-TA-TPGS的傅里叶红外光谱图
2.2 客体聚合物FA-AD的合成与分析
根据酰胺反应的基本原理,在EDC/NHS这对常用的氨基和羧基接枝催化试剂的加入下,合成了客体共聚物FA-AD[13]。简言之,先将FA和EDC/NHS溶解在含有20 ml CDDMSO的带塞锥形瓶中,并置于磁力搅拌器上搅拌1 h以活化羧基,然后再加入AD并在50 °C的温度下共同搅拌24 h,之后将获得的溶液倒入透析袋(Mr500 Da)中,并放置再去离子水中透析;最后,通过真空冷冻干燥获得客体聚合物FA-AD。同样采取核磁共振波谱仪和傅里叶变换红外光谱验证FA-AD的合成,结果见图3~4。图3所示,2.04(a)和1.57(b)处的特征质子峰分别归因于AD单元的(-CH2)3CH和(-CH2)(-NH2)CCH2-。8.70(c)、4.54(d)、6.99(e)、6.70(f)、7.70(g)、8.19(h)、4.38(i)、1.94(j)、2.36(k)和11.51(m)ppm处的峰分别归因于FA单元的-N=CH-、-CH2NH-、 -NHPh-、-Ph邻位氢、-Ph间氢、-PhCONH-、-CONHCHCOOH-、-CH2CH2COOH、-CH2COOH和-COOH。比较FA、AD和FA-AD的核磁共振氢谱可以看出,FA-AD几乎包含了FA和AD的所有特征峰。在FA-AD光谱中,属于FA的羧基质子峰(11.51 ppm)的消失,并且在8.07 ppm代表的酰胺基质子峰低于FA中的原始酰胺质子峰(8.19 ppm),这可能与引入AD引发的屏蔽效应有关。以上这些证明了FA-AD聚合物的成功合成。傅里叶变换红外光谱进一步证实了以上结论。如图4所示,FA在3120 cm-1处属于O-H的特征吸收峰未反映在FA-AD光谱中。此外,FA-AD光谱显示1696 cm-1(C=O),1608 cm-1(N-H)和1183 cm-1(C-N),表明存在酰胺键。相较之在FA光谱中,这些峰略有变化,这可能是由FA和AD反应形成新的酰胺键引起的。此外,在FA-AD光谱中,AD在2904 cm-1、2848 cm-1和1337 cm-1处的峰值略微偏移至2912 cm-1、2851 cm-1和1305 cm-1,表明FA与AD之间存在相互作用。因此,这些结果进一步证实了FA-AD的成功合成。
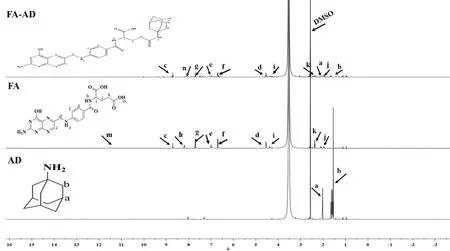
图3 AD,FA和FA-AD的核磁共振氢谱图
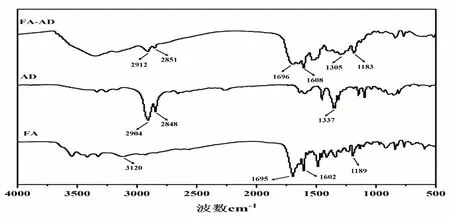
图4 AD,FA和FA-AD的傅里叶红外光谱图
2.3 CP-FA-RH NPs的制备
CP-FA-RH NPs的步骤主要分为以下三个步骤:第一步,称取RH 1 mg,CD-TA-TPGS 10 mg,分别溶解于1 ml DMSO溶液中,超声涡旋直至溶解,然后同时缓慢滴入装有20 ml纯水的烧杯中,反应1 h后,通过透析技术除去DMSO得到RH NPs溶液;第二步,称取2 mg FA-AD,同样溶于1 ml DMSO溶液中,超声涡旋并用0.8 μm微孔滤膜过滤,以得到清晰明亮的FA-AD溶液,然后,在超声处理条件下,将该溶液滴加到RH NPs溶液中,30 min后再将这溶液转移到透析袋中以除去DMSO得到FA-RH NPs溶液;最后一步,将壳聚糖溶液与果胶溶液以2:1的比例搅拌均匀后,滴入上一步制备的溶液中,然后再放置于磁力搅拌器上,匀速搅拌1 h后得到最终的CP-FARH NPs溶液。制备出的CP-FA-RH NPs溶液呈现澄清黄色状,无浑浊或沉淀析出,表明CP-FA-RH NPs溶液体系分布均一且稳定。对于每一步形成的NPs溶液,测定其平均粒径、PDI和Zeta电位,结果如表1所示(n=3)。相较于RH NPs,FA-RH NPs的粒径从110.93±2.39 nm增加到117.59±2.00 nm,Zeta电位从-11.2±0.3 mV降低到-13.1±0.3 mV,这些变化归因于主客体聚合物相互结合成功;另外,最终CP-FARH NPs粒径达到220.53±2.13 nm, Zeta电位逆转为20.6±1.8 mV,表明聚电解质CP在静电相互作用力下成功吸附在纳米粒子表面,证实了CP-FA-RH NPs的成功制备。此外,值得一提的是,在整个制备过程中,PDI值都很小,表明了制备的CP-FA-RH NPs粒径分布均一,稳定性好,从图5 CP-FA-RH NPs的粒径分布柱状图中也印证了这一点。
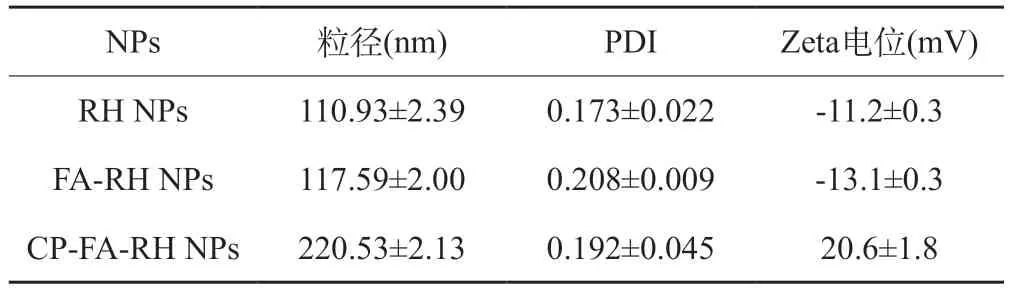
表1 每一步形成NPs的粒径,PDI和Zeta电位
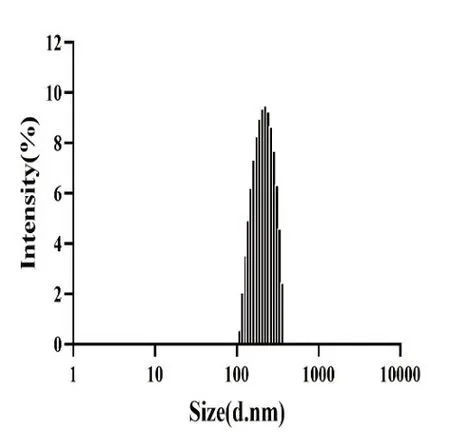
图5 CP-FA-RH NPs的粒径分布图
2.4 大黄酸含量测定
2.4.1 色谱条件 色谱柱为Comatex-C18柱(250 mm*4.6 mm, 5 μm);检测波长254 nm;流动相为甲醇-0.1%磷酸水(85:15);体积流量为1.0 mL·min-1;柱温为30 ℃;进样量为10 μl[14]。
2.4.2 对照品溶液的配制 精密称取RH对照品2.08 mg,置于100 ml容量瓶中,加入适量甲醇超声振荡溶解,再稀释定容至刻度线,配制成质量浓度为20.8 μg·mL-1的RH对照品溶液。分别精密吸取RH对照品溶液1 ml,750 μl,375 μl,250 μl,187.5 μl,125 μl,93.75 μl,62.5 μl置于10 ml容量瓶中,用甲醇定容至刻度,超声振荡,经0.22 μm的微孔滤膜过滤后加入棕色样品瓶中,按“2.4.1”项下色谱条件,进样10 μl测定峰面积,以RH对照品质量浓度x为横坐标,峰面积y为纵坐标绘制标准曲线(如图6),得到线性回归方程为y=27325x-821.14,R2=0.9995,表明大黄酸线性关系良好。
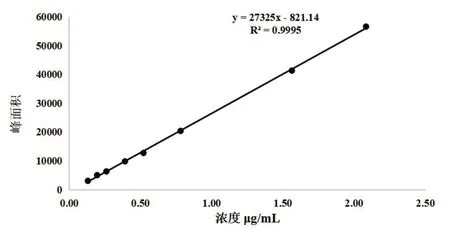
图6 RH含量测定标准曲线
2.4.3 CP-FA-RH NPs样品中大黄酸含量测定 精密量取1 ml CP-FA-RH NPs溶液置于25 mL容量瓶中,加入适量甲醇,超声破乳5 min,冷却至室温后,用甲醇定容至刻度线,用0.22 μm的微孔滤膜过滤后得样品溶液。按“2.4.1”项下色谱条件进样测定峰面积,通过线性回归方程计算出该样品中RH的质量浓度为1.21 μg·mL-1,并用以下公式计算出CP-FARH NPs样品中大黄酸的包封率(EE%)和载药量(LE%)[15]。
按以上公式计算出EE%为75.84%,LE%为5.06%,表明RH在CP-FA-RH NPs纳米体系中装载性能好,有效提高了RH的生物利用度,为该纳米制剂开发口服治疗疾病奠定了良好的基础。
2.5 CP-FA-RH NPs的微观形貌及X射线衍射图谱
2.5.1 CP-FA-RH NPs的微观形貌 量取1ml CP-FA-RH NPs溶液,用去离子水稀释至适当浓度,缓慢滴在具有碳支持膜的铜网上,停留5 min左右,用滤纸吸去多余液体,然后再用2%的磷钨酸溶液负染2 min,自然晾干后,在200 nm尺寸下用透射电子显微镜观察CP-FA-RH NPs的微观形貌。结果见图7,CP-FA-RH NPs呈圆球形,大小均匀,粒子之间无粘连,成型性好。

图7 CP-FA-RH NPs的微观形貌图
2.5.2 X射线衍射图谱 按照CP-FA-RH NPs的制备流程,在不加RH的条件下,制备空白纳米溶液(blank NPs),通过真空冷冻干燥获取blank NPs粉末和CPFA-RH NPs粉末。利用X射线衍射仪在40 kV、40 mA条件下,用5°/min的速度从5°扫描到90°,对RH粉末、所有材料混合物粉末,blank NPs粉末和CP-FARH NPs粉末进行晶型分析。结果如图8所示,RH图谱在15°至90°的范围内显示出许多尖锐的峰,表明了RH典型的晶体特性。相比之下,这些尖锐的峰没有出现在blank NPs和CP-FA-RH NPs中,这表明RH与所有载体基质之间没有形成任何结晶配合物,并以无定形状态成功包载在载体基质中。
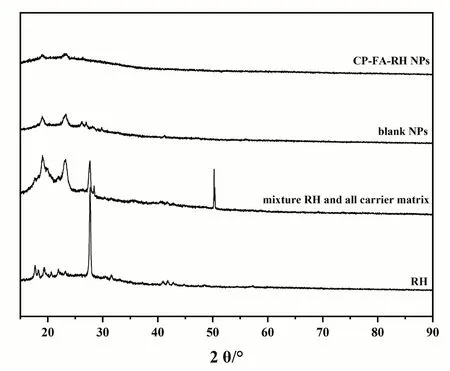
图8 X射线衍射图谱
2.6 巨噬细胞靶向性能研究
2.6.1 巨噬细胞培养 小鼠单核巨噬细胞白血病细胞(Raw 264.7)购自上海盖恩生物科技有限公司,在含有10%热灭活胎牛血清和1%青霉素链霉素的DMEM培养基中培养,所有细胞均置于37 ℃,5%CO2的细胞培养箱中孵育。
2.6.2 靶向性能评估 为了探究FA功能化的CP-FARH NPs的靶向能力,采用激光共聚焦显微镜对巨噬细胞摄取进行可视化图像分析。由于RH无荧光特性,因此利用疏水荧光探针香豆素6(C6)代替RH进行示踪。此外,基于口服纳米递药系统的构建,评估CP-FA-C6 NPs的靶向性能需要肠道菌群对CP层进行降解,然后再观察巨噬细胞对FA-C6 NPs的摄取[16]。因此,在没有肠道菌群相互作用的情况下,仅在FA-C6 NPs、C6 NPs和Free C6组进行靶向摄取分析,而在CP-FA-C6 NPs组中不进行分析。首先,将培养至对数生长期的Raw 264.7细胞接种于激光共聚焦小皿中,每皿接种约 1×105个细胞,然后放置于恒温培养箱培养 12 h;待细胞贴壁后,弃掉原培养基,分别加入含FA-C6 NPs、C6 NPs和Free C6的新鲜培养基(以C6计,浓度为 100 ng·mL-1)并继续放置于恒温培养箱中孵育;4 h后,再次弃掉皿中培养基,用冷PBS清洗3次,然后加入4%多聚甲醛固定10 min,再用冷PBS清洗3次后,加入1 ml DAPI溶液,10 min后再次吸取冷PBS进行清洗;最后,加入抗荧光猝灭剂,将小皿放置于激光共聚焦显微镜下进行观察。蓝色荧光代表细胞核,绿色荧光代表C6标记的NPs。结果如图9所示,在Free C6组中观察到较差的绿色荧光,弱于C6 NPs组和FA-C6 NPs组;其中,FA-C6 NPs组观察到的荧光最亮,表明FA功能化的NPs更容易被Raw 264.7细胞摄取。为了确认由巨噬细胞表面FA受体介导的FA功能化NPs的靶向摄取,采用游离FA溶液预处理细胞1h,然后再分别与C6 NPs和FA-C6 NPs一起孵育4 h。结果表明,与未使用游离FA溶液预处理的FA-C6 NPs组相比,游离FA溶液预处理后FA-C6 NPs组细胞的荧光强度显著降低。然而,在C6 NPs组中没有发现类似的变化,进一步阐明了FA-C6 NPs通过FA介导的内吞作用特异性被Raw 264.7细胞靶向摄取。结合以上结果,本文研究设计制备的CP-FA-RH NPs具备靶向巨噬细胞的能力,为以后开发或借鉴用于靶向治疗提供了良好的基础。

图9 巨噬细胞对不同纳米制剂靶向摄取图
3 讨论
早些年前,国内外学者就在超分子诊疗理念中指出了基于超分子聚合物构建纳米递药系统的可行性和有效性[17]。超分子纳米递药系统可以在水溶液和盐溶液中保持完整的结构。在过去几年的研究中,很多超分子纳米载药平台在癌症治疗领域取得了突破性进展,其中一些已获准用于临床癌症治疗,如Doxil(聚乙二醇化脂质体阿霉素)、Onivyde(伊立替康脂质体)和Abraxane(白蛋白结合紫杉醇)等[18-20]。本研究开发合成聚合物CD-TA-TPGS,结合主客体相互作用力和静电相互作用力,逐步将FA-AD和CP吸附在其表面,最终成功制备出了具备口服靶向巨噬细胞的大黄酸超分子纳米粒CP-FA-RH NPs,它不仅有效解决了RH溶解度低,生物利用度差的问题,还有效实现了RH的靶向递送,大大减少了其在疾病治疗上对健康组织带来的损伤,有望满足日益增长的医疗需求。然而,尽管如此,在应对复杂的病理环境,超分子纳米靶向递药系统的性能优化依然成为临床治疗发展的重中之重,对于载药体系的生物稳定性、安全性、体内分布和代谢途径等等的系统研究迫在眉睫,而考察和评价其在不同层面的作用和治疗效果并逐步走向临床也尚需时日。
