蒙药材山苦荬的成分鉴别及7种成分含量测定方法建立Δ
2024-01-03孙丽君王秋桐夏慧敏王跃武张慧文王焕芸内蒙古医科大学药学院呼和浩特000内蒙古自治区新药筛选工程研究中心呼和浩特000内蒙古医科大学附属医院药物临床试验机构呼和浩特000
孙丽君 ,李 君 ,王秋桐 ,夏慧敏 ,王跃武 ,张 谦 ,张慧文 ,王焕芸 #(.内蒙古医科大学药学院,呼和浩特 000;2.内蒙古自治区新药筛选工程研究中心,呼和浩特 000;.内蒙古医科大学附属医院药物临床试验机构,呼和浩特 000)
山苦荬为菊科苦荬菜属多年生植物山苦荬Ixeris chinensis(Thunb.)Nakai的干燥全草,蒙医称之为“苏素-乌布斯”,现收录于《内蒙古蒙药材标准》。该药性寒、味苦,具有清血热、抗炎、抗氧化、保肝等功效,常用于蒙医临床肝炎、肝胆热等疾病的治疗[1]。研究表明,山苦荬中所含绿原酸、槲皮素具有协同抗炎作用[2];原儿茶酸可通过调控Toll样受体4/髓样分化因子88/核因子κB信号通路而达到改善肝脏炎症的目的[3];在四氯化碳诱导的肝损伤小鼠模型中,木犀草素可显著降低其血清中谷草转氨酶、谷丙转氨酶的活性,并改善肝组织病理损伤,作用机制可能与激活核转录因子红系2 相关因子2 通路有关[4];异绿原酸A、芦丁可通过增强抗氧化酶活性而抑制氧化应激反应,从而发挥保肝作用[5-6]。同时,课题组前期在山苦荬保肝活性部位筛选过程中发现,木犀草苷具有显著的保肝活性[1]。
药材所含成分含量的高低与其疗效密切相关,虽有学者采用高效液相色谱法和一测多评法对山苦荬药材进行了质量评价[7-8],但受地域差异等因素影响,仅以单一或3种成分作为质量评价指标难以准确反映药材整体质量,加之目前关于山苦荬的物质基础研究有限,因此明确山苦荬的物质组成并进行质量控制对保障其临床应用的安全性和有效性具有良好的现实意义。基于此,本研究拟运用高效液相色谱-四极杆/静电场轨道阱高分辨质谱技术对山苦荬药材的物质组成进行快速、全面的鉴别分析,并采用高效液相色谱串联三重四极杆质谱(high performance liquid chromatography-triple quadrupole mass spectrometry,HPLC-MS/MS)技术对其所含绿原酸、木犀草素、槲皮素、芦丁、原儿茶酸、异绿原酸A、木犀草苷7种成分进行含量测定,旨在为该药材质量评价体系的建立、药效物质的挖掘提供数据支撑。
1 材料
1.1 主要仪器
本研究所用主要仪器有HPLC-Q-Exactive型高效液相色谱-四极杆/静电场轨道阱高分辨质谱仪[赛默飞世尔科技(中国)有限公司],LC-MS8045型高效液相色谱-三重四极杆液质联用仪、AP135W型十万分之一电子天平(日本Shimadzu公司)等。
1.2 主要药品与试剂
异绿原酸A对照品(批号PS001052,纯度≥98.0%)、隐绿原酸对照品(批号PS011467,纯度≥98.0%)、木犀草苷对照品(批号PS013155,纯度≥98.0%)、木犀草素对照品(批号PS010346,纯度≥98.0%)均购自成都普思生物科技有限公司;绿原酸对照品(批号110753-202119,纯度≥98.5%)、槲皮素对照品(批号100081-202010,纯度≥99.0%)、原儿茶酸对照品(批号110809-202207,纯度≥98.0%)、枸橼酸对照品(批号100396-202104,纯度≥99.7%)、阿魏酸对照品(批号110773-201915,纯度≥99.0%)、芦丁对照品(批号100080-202012,纯度≥98.0%)均购自中国食品药品检定研究院;甲醇、乙腈均为色谱纯,甲酸均为分析纯,水为蒸馏水。
山苦荬药材(批号分别为C20210514、T20210607、H20210523)分别产自内蒙古赤峰市、通辽市、呼和浩特市,经内蒙古医科大学药学院中药资源教研室渠弼教授鉴定为菊科苦荬菜属植物山苦荬I.chinensis(Thunb.)Nakai的干燥全草。
2 方法与结果
2.1 山苦荬的成分鉴别
2.1.1 溶液的制备
(1)混合对照品溶液:精密称定绿原酸对照品5.03 mg、枸橼酸对照品4.12 mg、隐绿原酸对照品4.95 mg、阿魏酸对照品3.97 mg、木犀草素对照品4.20 mg、槲皮素对照品5.05 mg、芦丁对照品4.01 mg、原儿茶酸对照品5.02 mg、异绿原酸A 对照品7.50 mg、木犀草苷对照品3.92 mg 分别置于10 mL 容量瓶中,甲醇溶解、超声、定容,得单一对照品贮备液,质量浓度分别为503.00、412.00、495 .00、397.00、420.00、505.00、401.00、502.00、750.00、392.00 μg/mL;分别精密量取上述各单一贮备液100 μL于同一10 mL容量瓶中,得质量浓度分别为5.03、4.12、4.95、3.97、4.20、5.05、4.01、5.02、7.50、3.92 μg/mL的混合对照品溶液,于4 ℃下保存,备用。
(2)供试品溶液:取山苦荬药材(批号C20210514)粉末(过三号筛)约1.0 g,精密称定,放至50 mL锥形瓶中,加甲醇10 mL,称重,封口,超声(功率200 W,频率50 kHz)提取30 min,冷却至室温,再次称重,用甲醇补足失重,摇匀,经0.22 μm滤膜过滤,即得。
2.1.2 色谱与质谱条件
(1)色谱条件:以Agilent ZORBAX SB-Aq(4.6 mm×150 mm,5 μm)为色谱柱,以甲醇(A)-0.1%甲酸溶液(B)为流动相进行梯度洗脱(0~10 min,10%A;10~13 min,10%A→28%A;13~15 min,28%A→45%A;15~21 min,45%A→65%A;21~24 min,65%A→80%A;24~34 min,80%A→95%A;34~40 min,95%A),流速为0.4 mL/min,柱温为35 ℃,进样量为5 μL。
(2)质谱条件:采用电喷雾离子源(electron spray ionization,ESI)进行正、负离子扫描;喷雾电压为3.80 kV(正离子)/3.20 kV(负离子);离子传输管温度为300 ℃(正离子)/400 ℃(负离子);辅助气体积流量为30 L/min;辅助气温度为350 ℃;碰撞能量设置为30 eV;检测方式为Full MS/dd-MS2,Full MS 分辨率为70 000,dd-MS2分辨率为17 500;扫描范围为m/z110~1 200。
2.1.3 数据库建立与分析
采用中医药系统药理学数据库与分析平台(https://old.tcmsp-e.com/tcmsp.php)、中国知网、万方数据对山苦荬药材所含成分的名称、分子式、多级质谱碎片进行收集整理,建立本地数据库,利用Thermo Scientific Xcalibur 3.0软件,对山苦荬供试品溶液和混合对照品溶液在正、负离子模式下的总离子流数据进行处理,并与本地自建数据库比对,以一级质谱误差<10 ppm为标准对山苦荬中的成分进行指认[9],并将各成分元素组成、多级质谱碎片信息及与现有对照品比对作进一步确认,最终鉴定山苦荬中化学成分。
2.1.4 山苦荬药材化学成分鉴定
取“2.1.1”项下溶液,按“2.1.2”项下色谱与质谱条件进样分析,得山苦荬供试品溶液和混合对照品溶液正、负离子模式下的总离子流图(图1、图2)。结果显示,从山苦荬中共鉴定出45 个化学成分,包括20 个有机酸类成分、13 个黄酮类成分、4 个氨基酸类成分、4 个脂肪酸类成分、3 个核苷类成分、1 个香豆素类成分,其中10 个成分经与对照品比对得到进一步确证(表1)。

表1 山苦荬中化学成分的分析结果

图1 供试品溶液总离子流图

图2 混合对照品溶液总离子流图
以有机酸类为例,山苦荬中20个有机酸类化合物被鉴定。该类化合物是由咖啡酸与奎宁酸通过酯化缩合产生,酯键断裂可产生奎宁酸特征碎片m/z191 和咖啡酸特征碎片m/z179;咖啡酸脱羧后产生特征碎片m/z135,依此可作为判断此类化合物的裂解规律[15]。以异绿原酸A(化合物30)为例,其在负离子模式下的准分子离子峰为m/z515.119 51[M-H]-,Thermo Scientific Xcalibur 3.0软件拟合其分子式为C25H24O12,误差为2.150 ppm;主要碎片离子峰为m/z353.088 23、191.055 48、179.034 16、135.044 01,即准分子离子峰经高能撞击后,首先失去1分子咖啡酸基团产生m/z353.088 23[M-H-C9H6O3]-,进一步丢失1 分子咖啡酸基团产生m/z191.055 48[M-H-C9H6O3-C9H6O3]-,或丢失1分子奎宁酸基团产生m/z179.034 16[M-H-C9H6O3-C7H10O5]-碎片离子峰,后者进一步脱去1分子CO2产生m/z135.044 01[MH-C9H6O3-C7H10O5-CO2]-。通过分子式、化合物裂解规律、相关文献[14]及与对照品比对,确认30 号化合物为异绿原酸A。化合物30 可能的裂解途径及二级质谱图见图3。
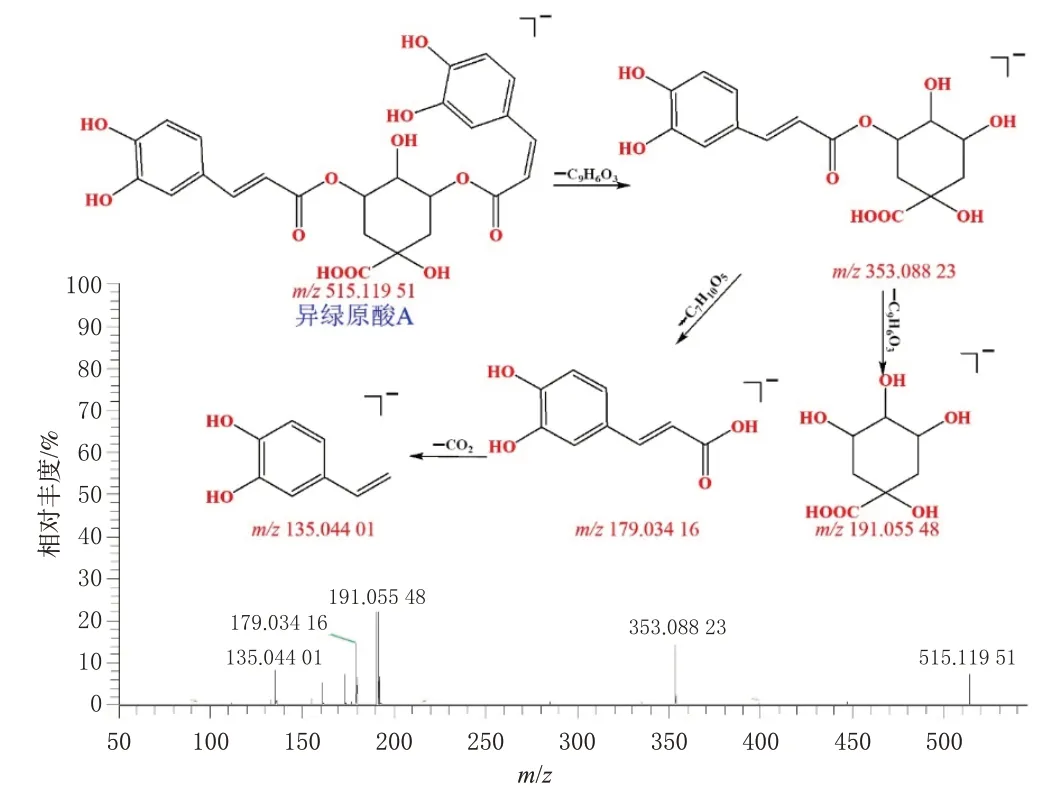
图3 化合物30可能的裂解途径及MS2图
2.2 山苦荬中7种成分的含量测定
2.2.1 溶液的制备
(1)混合对照品溶液的制备:精密称定绿原酸对照品12.58 mg、异绿原酸A 对照品5.25 mg、木犀草苷对照品9.80 mg 对照品置于同一10 mL 容量瓶中,精密量取“2.1.1”项下单一对照品贮备液木犀草素1 000 μL、槲皮素10 μL、芦丁200 μL、原儿茶酸100 μL 置于上述同一10 mL容量瓶中,甲醇溶解、定容,得绿原酸、木犀草素、槲皮素、芦丁、原儿茶酸、异绿原酸A、木犀草苷质量浓度分别为1 258.00、42.00、0.51、8.02、5.02、525.00、980.00 μg/mL 的混合对照品溶液,于4 ℃保存,加样回收率试验备用。
(2)供试品溶液的制备:按“2.1.1(2)”项下方法制备,摇匀,取1 mL溶液,置于10 mL容量瓶中,甲醇定容,摇匀,经0.22 μm滤膜过滤,即得。
(3)空白对照溶液的制备:取甲醇适量,按“2.1.1(2)”项下供试品溶液制备方法处理,即得。
2.2.2 色谱与质谱条件
(1)色谱条件:以Shim-pack GIST-HP C18(2.1 mm×100 mm,3 μm)为色谱柱,以甲醇(A)-0.1%甲酸溶液(B)为流动相进行梯度洗脱(0.01~0.3 min,10%A→23%A;0.3~3.0 min,23%A→45%A;3.0~4.3 min,45%A→65%A;4.3~5.5 min,65%A→95%A;5.5~7.0 min,95%A;7.0~7.01 min,95%A→10%A;7.01~9.5 min,10%A);流速为0.25 mL/min,柱温为35 ℃,进样量为3 μL。
(2)质谱条件:采用ESI,以多反应监测(multiple reaction monitoring,MRM)模式进行负离子扫描;脱溶剂温度为526 ℃;雾化气流量为3 L/min;加热气流量为10 L/min。7个待测成分的质谱参数见表2,混合对照品溶液、供试品溶液图谱见图4(空白对照溶液图谱略)。
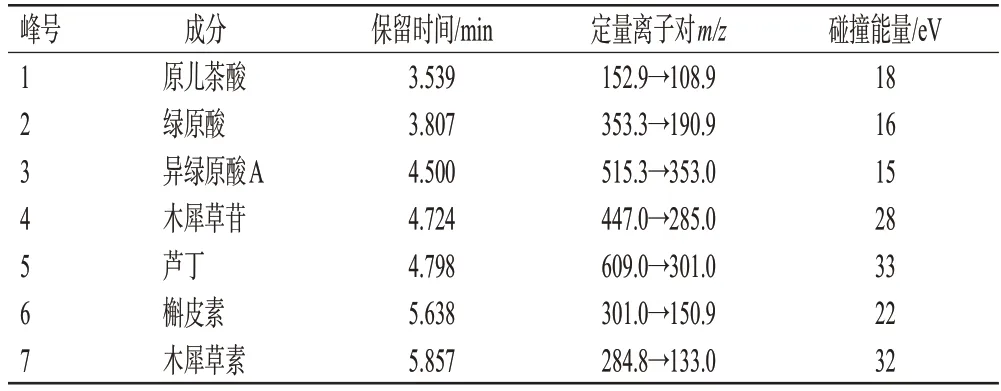
表2 7种成分的保留时间及质谱分析参数
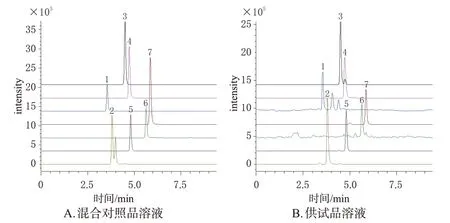
图4 7种成分的MRM色谱图
2.2.3 线性关系考察
精密量取“2.1.1(1)”项下绿原酸、木犀草素、槲皮素、芦丁、原儿茶酸、异绿原酸A、木犀草苷单一对照品贮备液适量于同一10 mL容量瓶中,甲醇稀释、定容,得质量浓度为50.300、4.200、0.505、2.005、2.510、75.000、19.600 μg/mL的混合对照品溶液,分别精密吸取上述混合对照品溶液10、20、50、100、200、400、500 μL,用甲醇分别定容至1 mL,制备系列质量浓度混合对照品溶液,按“2.2.2”项下色谱与质谱条件进样分析,分别以各成分质量浓度(X)为横坐标,峰面积(Y)为纵坐标,通过HPLC-MS/MS联用系统中LabSolutions软件对各待测成分进行线性拟合,结果见表3。

表3 山苦荬中7种成分的回归方程与线性范围
2.2.4 精密度试验
取“2.2.3”项下混合对照品溶液(绿原酸、木犀草素、槲皮素、芦丁、原儿茶酸、异绿原酸A、木犀草苷质量浓度分别为2 515.00、210.00、25.25、100.25、125.50、3 750.00、980.00 ng/mL),按“2.2.2”项下色谱与质谱条件连续进样6 次,记录峰面积。结果显示,各成分峰面积RSD均小于3.00%(n=6),表明仪器精密度良好。
2.2.5 稳定性试验
精密称定山苦荬药材粉末(批号C20210514)约1.0 g,精密称定,按“2.2.1(2)”项下方法制备供试品溶液,分别于室温下放置0、3、6、9、12、24 h时按“2.2.2”项下色谱与质谱条件进样分析,记录峰面积。结果显示,各成分峰面积RSD均小于3.00%(n=6),表明供试品溶液在室温放置24 h内稳定性良好。
2.2.6 重复性试验
精密称定山苦荬药材粉末(批号C20210514)约1.0 g,平行6 份,按“2.2.1(2)”项下方法制备供试品溶液,再按“2.2.2”项下色谱与质谱条件进样分析,记录峰面积并代入回归方程计算样品含量。结果显示,绿原酸、木犀草素、槲皮素、芦丁、原儿茶酸、异绿原酸A、木犀草苷平均含量分别为2 582.90、97.73、0.95、16.52、9.35、1 031.87、1 919.05 μg/g,且RSD均小于3.00%(n=6),表明方法重复性良好。
2.2.7 加样回收率试验
精密称定已知含量的山苦荬药材粉末(批号C20210514)6 份,每份约0.5 g,精密称定,分别加入“2.2.1(1)”项下混合对照品溶液适量(加入量与已知量相等),按“2.2.1(2)”项下方法制备供试品溶液,再按“2.2.2”项下色谱与质谱条件进样分析,记录峰面积并计算加样回收率。结果显示,绿原酸、木犀草素、槲皮素、芦丁、原儿茶酸、异绿原酸A、木犀草苷的平均回收率分别为101.48%、99.83%、99.35%、96.72%、105.84%、98.29%、99.58%(RSD 分别为2.24%、2.94%、3.22%、3.16%、3.07%、2.60%、2.44%,n=6)。
2.2.8 样品含量测定
取3 批山苦荬药材(H20210523、T20210607、C20210514)各3 份,按“2.2.1(2)”项下方法制备供试品溶液,再按“2.2.2”项下色谱与质谱条件进样测定,记录峰面积并代入回归方程计算样品含量,结果见表4。

表4 不同来源山苦荬中7种成分平均含量(n=3,μg/g)
3 讨论
3.1 样品前处理条件及成分鉴别的色谱和质谱条件优化
本课题组前期对样品处理方式(超声处理、加热回流处理)、处理时间(15、30、45 min)进行考察,结果显示,超声处理30 min 的提取效率为最优。本课题组前期对不同流动相体系进行比较发现,相比于乙腈,以甲醇作为有机相时各成分的分离度更好;同时,在水相中加入0.1%甲酸能有效提高各成分的质谱响应并改善色谱峰峰形,故最终将流动相体系确定甲醇-0.1%甲酸溶液(梯度洗脱)。传统药材成分组成复杂多样,且各成分在不同电离模式下离子化程度和响应强度均有所差异,为全面、精准表征山苦荬药材所含化学成分,本研究采用正、负两种电离模式对山苦荬药材中的化学成分进行辨识分析。
3.2 含量测定的质谱条件优化
传统药材饮片中所含成分含量的高低与其药效的强弱、质量的优劣密切相关。为保障山苦荬药效和质量的稳定性,本研究通过HPLC-MS/MS 技术对其中的绿原酸、木犀草素、槲皮素、芦丁、原儿茶酸、异绿原酸A、木犀草苷7种成分进行了含量测定。本课题组前期采用质谱全扫描方式对各待测成分的离子化模式进行了确认,结果显示,7种成分在负离子模式下的响应均优于正离子模式;在确认各待测成分的离子化模式后,本课题组通过设置不同碰撞能量分别进行产物离子扫描,初步获得碰撞能量、前体离子及产物离子后,采用质谱自动优化程序对碰撞能量、前体离子及产物离子进一步优化,最终获得各待测成分的质谱分析参数。
3.3 含量测定的结果分析
3 批不同来源山苦荬药材含量测定结果表明,批号C20210514 和批号T20210607 的山苦荬药材中槲皮素、芦丁及原儿茶酸含量均明显低于批号H20210523样品,而其他各成分含量均明显高于批号H20210523样品;与批号T20210607 样品相比,批号C20210514 的山苦荬药材中芦丁含量较高,而绿原酸含量较低,其他成分含量则相当。由此可见,地域差异对山苦荬药材中上述成分的含量存在一定影响,但由于本研究涉及样本来源较为局限,后期将扩大药材收集范围,作进一步评价。
综上所述,本研究所建成分鉴别和含量测定方法快速、简便,可为山苦荬质量标准建立提供科学依据,也可为今后山苦荬血浆移行成分解析、移行-网络分析奠定数据基础。
