无定形Fe基MOF材料的制备及析氧性能研究
2024-01-03秦王昕上官向阳王康军于广莉
秦王昕, 上官向阳, 王康军, 于广莉
(沈阳化工大学 化学工程学院, 辽宁 沈阳 110142)
随着经济与科学技术的快速发展,能源危机和环境恶化问题日益严峻,因此清洁能源得到了广泛的需求和推广[1-2].在众多可替代能源中,氢能(H2)因来源广、能量密度高等优点被认为是一种有前途的可再生能源[3],并且H2是一种零碳排的能源载体,水是唯一燃烧产物,因而开发利用H2是实现2050年碳中和目标的有效途径.电解水是生产高纯H2的有效方法之一,但阳极析氧反应(OER)涉及复杂4e-转移过程,需要较大的过电位才能克服动力学迟缓问题,从而限制了整个电解水制氢的效率[4-5].目前,RuO2、IrO2等贵金属氧化物对OER具有优异的电催化性能,但其固有的金属稀缺性、高昂的成本阻碍了其大规模应用.因此,开发廉价、高效的OER催化剂仍然是一项巨大挑战.
晶态金属有机框架(MOF)是由无机节点(金属离子或簇)与有机配体通过配位键自组装而形成的一类新型多孔材料.因其高度有序的多孔性、大的比表面积和可调节的金属活性中心等优势,MOF在气体储存/分离、催化、药物缓释和传感等方面具有潜在的应用前景[6-12].近年来,具有过渡金属中心的MOF材料(如CoFe-MOF、FeNi-MOF和NiCo-MOF等)在电催化领域取得了初步进展[13-15].然而,MOF良好的结晶度是一把双刃剑,多数活性位点被有机配体包裹而难以得到有效利用,大大降低了其催化活性.与晶态MOF材料相比,无定形MOF(aMOF)保留了晶态MOF的基本结构单元和连通性,缺乏长程有序的网格结构,已逐渐引起科研工作者的关注.aMOF材料不仅具有较高的热、化学稳定性,而且金属中心的配位数较低,大量的缺陷促使材料表面以及内部暴露出更多的活性位点.迄今为止,aMOF材料已初步应用于药物缓释和气体吸附等研究领域[16-18],在电催化方面的研究较少.
笔者采用外来金属离子诱导策略,以均苯三甲酸为有机配体制备了一系列无定形的InxFey-aMOF材料.研究发现:Fe3+扰乱了In3+与有机配体配位,致使原始晶态In-MOF失去长程有序结构而暴露出更多的本征活性位点;Fe-aMOF表现出最佳的电催化性能,电流密度为0.01 A/cm2时的过电位仅为258.2 mV,塔菲尔斜率为45.4 mV/dec,优于大多数传统的晶态MOF材料;同时该催化剂还具有优异的长期稳定性.
1 实验部分
1.1 实验试剂
九水合硝酸铁[Fe(NO3)3·9H2O,分析纯]、水合硝酸铟[In(NO3)3·xH2O,质量分数为99%]、1,3,5-均苯三甲酸(H3BTC,质量分数为98%)和氢氧化钾(KOH,质量分数为99%),上海阿拉丁生化科技股份有限公司;导电碳黑(VXC-72R),卡博特有限公司;Nafion溶液(质量分数为5%),上海兢翀电子科技发展有限公司;泡沫镍(NF,厚1.0 mm),昆山广嘉源新材料有限公司;无水乙醇(C2H5OH,分析纯)、盐酸(HCl,质量分数为36%~38%)、丙酮(C3H6O,分析纯)和N,N-二甲基甲酰胺(DMF,分析纯),国药集团化学试剂有限公司.
1.2 InxFey-aMOF的制备
In1Fe1-aMOF的制备过程如下:首先,将450 mg(1.50 mmol)Fe(NO3)3·9H2O和604 mg(1.50 mmol)In(NO3)3·xH2O先后溶解于10 mL DMF和40 mL水的混合溶液中,搅拌15 min后,加入660 mg(3.14 mmol)1,3,5-均苯三甲酸,继续搅拌20 min形成均一混合液;其次,将上述混合液转移至100 mL高压反应釜中,并在120 ℃烘箱中晶化5 d;再其次,混合液冷却至室温,用离心方法(10 000 r/min,10 min)收集固体粉末,依次用DMF、水、乙醇将粉末洗涤3次,除去未反应杂质;最后,将得到的产物在100 ℃真空烘箱中干燥8 h,命名为In1Fe1-aMOF.
金属离子总物质的量保持为3.0 mmol,通过改变Fe(NO3)3·9H2O和In(NO3)3·xH2O初始物质的量的比来调控产物中金属的组分,即In、Fe的初始物质的量的比为1∶0、1∶3、1∶4和0∶1,相应产物分别命名为In-MOF、In1Fe3-aMOF、In1Fe4-aMOF和Fe-aMOF,其制备方法与In1Fe1-aMOF相同.
1.3 实验仪器
采用Rigaku SmartLab X射线衍射仪表征电催化剂的晶体结构(λ=0.154 18 nm,40 kV,30 mA),扫描范围为4°~40°,扫描速率为10 (°)/min;采用Nicolet iS50傅里叶变换红外光谱仪对电催化剂的官能团进行测试,测试范围为400~4 000 cm-1;采用蔡司ZEISS Gemini 300扫描电子显微镜观察催化剂的形貌、尺寸和元素分布;采用FEI TECNAI G2F20透射电子显微镜分析催化剂的结构、有序性等微观信息;采用康塔Autosorb-IQ-C全自动物理吸附仪表征催化剂的比表面积和孔结构,测试前样品在120 ℃下真空活化10 h;采用Thermo Fisher iCAP 7400电感耦合等离子体原子发射光谱仪对催化剂的组成进行定量测试.所有催化剂的电化学性质均在上海辰华CHI 600E型电化学工作站上进行.
1.4 电化学性能测试
1.4.1 泡沫镍的预处理
首先,将商用泡沫镍(NF)剪成1 cm × 2 cm,在丙酮中超声清洗3次,去除表面油污;其次,在1 mol/L盐酸溶液中超声清洗3次,去除表面氧化物;再其次,用去离子水超声清洗,直至溶液呈中性;最后,用无水乙醇超声清洗3次,将洗涤干净的NF在60 ℃真空烘箱中干燥2 h,备用.
1.4.2 工作电极的制备
分别称取5 mg催化剂和5 mg碳粉置于1.5 mL离心管中,再依次加入440 μL无水乙醇、60 μL Nafion溶液.将上述混合物超声至糊状.移取80 μL糊状混合物均匀涂抹在导电载体NF上(涂覆面积约为1 cm × 0.8 cm),室温下晾干,备用.
1.4.3 析氧反应性能测试
选用典型的三电极体系,将1.4.2所制备的电极作为工作电极,铂片作为辅助电极,Ag/AgCl作为参比电极,测试温度为25 ℃,电解液为1 mol/L的KOH.根据能斯特方程:ERHE=EAg/AgCl+0.059 2pH+0.197,将所有电位转换为可逆氢电极(RHE).评估OER活性前,在0.1~0.6 V(vs.Ag/AgCl)电位窗口进行50圈循环伏安(CV)扫描,使催化剂活化.线性扫描伏安曲线(LSV)测试条件:扫描速率为5 mV/s;电势窗口为0.9 ~1.6 V(vs.RHE).由LSV曲线计算可得塔菲尔(Tafel)斜率:η=a+blogJ(a为反应常数;b为Tafel斜率;J为电流密度).电化学阻抗谱(EIS)是在电压1.46 V(vs.RHE)、频率为100 kHz ~ 0.01 Hz、振幅为5 mV条件下获得.通过双层电容(Cdl)评估催化剂的电化学活性表面积(ECSA),在20、40、60、80、100 mV/s扫描速率下对非法拉第区域进行CV测试,利用0.125 V处的电流密度差(ΔJ/2)与扫描速率作图,斜率即为Cdl值.采用计时电流法,在电流密度为0.01 A/cm2的条件下测定催化剂的长期稳定性.
2 结果与讨论
2.1 基本表征
利用X射线衍射技术(XRD)表征In-MOF、In1Fe1-aMOF、In1Fe3-aMOF、In1Fe4-aMOF和Fe-aMOF的晶体结构,结果见图1.

图1 拟合的CPM-5材料、In-MOF、In1Fe1-aMOF、In1Fe3-aMOF、In1Fe4-aMOF和Fe-aMOF的XRD图谱
由图1可以看出:In-MOF衍射峰位置与单晶模拟的CPM-5[19]衍射峰位置相匹配,表明他们具有相同的晶体结构.In1Fe1-aMOF、In1Fe3-aMOF、In1Fe4-aMOF和Fe-aMOF的谱图中仅出现几个宽峰,并随着In、Fe初始物质的量的比减小,峰强度也逐渐降低,说明样品的结晶度较差.这可能是由于Fe3+的引入改变了局域电子分布,导致原始晶态In-MOF失去长程有序结构.
为了进一步探究催化剂的结构,采用红外光谱仪对H3BTC、In-MOF、In1Fe1-aMOF、In1Fe3-aMOF、In1Fe4-aMOF和Fe-aMOF的官能团进行测定,结果如图2所示.在H3BTC的红外光谱图中,1 723 cm-1和1 408 cm-1处的吸附峰归属于羧基(—COOH)伸缩振动.在In-MOF、In1Fe1-aMOF、In1Fe3-aMOF、In1Fe4-aMOF和Fe-aMOF的红外光谱图中,1 622 cm-1和1 372 cm-1处的吸附峰归属于去质子化羧基(—COO-)不对称和对称伸缩振动,同时在473 cm-1处检测到新的振动吸收峰,表明金属离子(In3+和Fe3+)与有机配体之间成功配位[20].通过ICP-AES测试定量分析催化剂中In3+和Fe3+的含量(见表1),随着Fe(NO3)3·9H2O物质的量的增加,骨架中n(In)/n(Fe)比值逐渐降低.此外,In1Fe1-aMOF、In1Fe3-aMOF和In1Fe4-aMOF中n(In)∶n(Fe)分别为1∶1.2、1∶3.8、1∶5.1,略大于初始投料比,表明Fe3+与H3BTC间的配位能力更强,部分In3+并未参与配位而留在母液中.

图2 H3BTC、In-MOF、In1Fe1-aMOF、In1Fe3-aMOF、In1Fe4-aMOF和Fe-aMOF的红外光谱
通过扫描电子显微镜(SEM)和透射电子显微镜(TEM)对Fe-aMOF的微观结构进行表征,结果如图3所示.由图3(a)可以看出Fe-aMOF呈球形,颗粒大小均匀,尺寸约为800 nm.由图3(c)—图3(e)可以看出:样品中含有Fe、C和O三种元素,在整个材料中均匀分布,进一步证明Fe3+与有机配体之间成功配位,且催化活性位点分散良好.通过EDS分析可知Fe的质量分数约为50%.

图3 Fe-aMOF的SEM图、EDS光谱及其相应的元素分布、TEM图和SAED图
在高分辨TEM图像[图3(f)]和相应的选定区域SAED[图3(g)]中并未观察到明显的晶格条纹和可见的衍射环,进一步证明Fe-aMOF具有非晶态特征,与XRD结果相一致.由于非晶材料富含大量的缺陷位点,促使Fe-aMOF拥有更高的本征活性,有利于提高其电催化性能.
采用N2物理吸附探究In-MOF和Fe-aMOF的多孔结构,结果如图4所示.由图4可以看出:In-MOF和Fe-aMOF在低压区快速吸附N2,说明两种材料都存在微孔;与In-MOF相比,Fe-aMOF在高压区平缓吸附N2,说明Fe-aMOF结构中还有部分介孔.图4(a)显示In-MOF的孔径分布比较窄,主要集中于0.54~0.93 nm.而Fe-aMOF具有较宽的孔径分布区间,除微孔外(0.52~2.00 nm),在2.00~10.00 nm间还出现了大量介孔[见图4(b)],这进一步证明了Fe3+导致局部配位缺失,形成微-介孔结构.In-MOF和Fe-aMOF的孔结构信息见表2.Fe-aMOF和In-MOF的比表面积分别为506 m2/g和670 m2/g,Fe-aMOF的介孔孔容(0.190 cm3/g)远高于In-MOF的介孔孔容(0.072 cm3/g),表明Fe-aMOF骨架中存在较多的介孔.这种微-介孔结构有利于提高活性比表面积和促进物质传输,从而提高Fe-aMOF的电催化性能.

表2 In-MOF和Fe-aMOF在77 K时的N2吸附结果
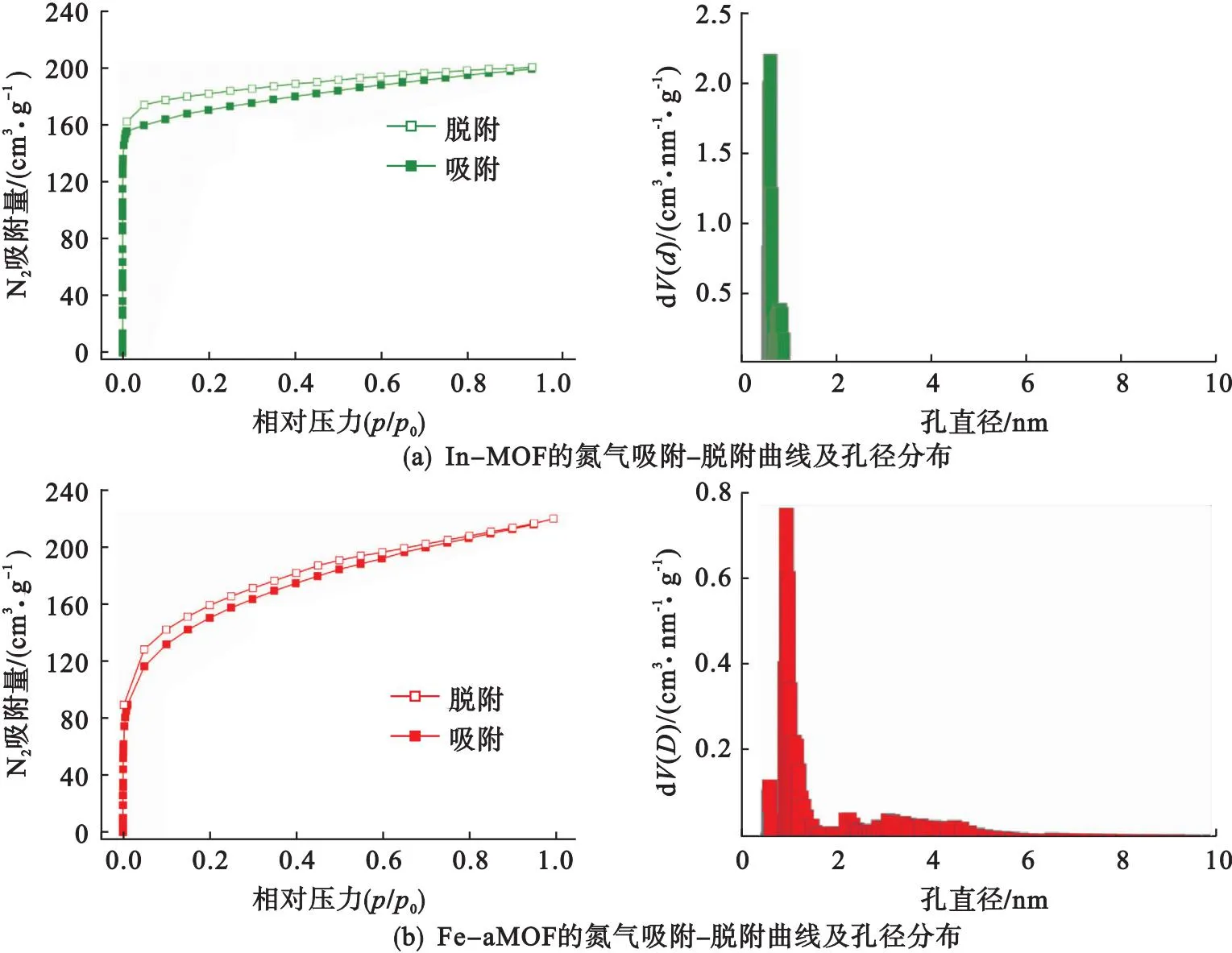
图4 In-MOF和Fe-aMOF的氮气吸附-脱附曲线及其相应的孔径分布
2.2 电化学性能测试
将In-MOF、In1Fe1-aMOF、In1Fe3-aMOF、In1Fe4-aMOF和Fe-aMOF制成工作电极,采用典型的三电极体系,在1.0 mol/L的KOH溶液中探究其OER性能,结果如图5所示.由图5(a)可以看出:In-MOF在0.1 A/cm2时的过电位为341.9 mV;通过外来金属离子诱导策略,随着Fe3+含量的增加,OER性能也逐渐增强.Fe-aMOF表现出最高的OER活性,起始电位和过电位都很低:在电流密度为0.01 A/cm2时的过电位仅为258.2 mV,明显低于In1Fe1-aMOF(301.1 mV)、In1Fe3-aMOF(279.6 mV)、In1Fe4-aMOF(269.8 mV)和空白NF(373.9 mV)的过电位;Fe-aMOF的过电位还低于大多数Fe基MOF催化剂[21-31]的过电位(见表3).为了深入研究电极材料的OER反应动力学,通过上述极化曲线,利用Tafel方程计算得出Tafel斜率.如图5(b)所示,Fe-aMOF的Tafel斜率为45.4 mV/dec,低于In-MOF(68.8 mV/dec)、In1Fe1-aMOF(48.3 mV/dec)、In1Fe3-aMOF(46.9 mV/dec)、In1Fe4-aMOF(46.0 mV/dec)和空白NF(105.4 mV/dec)的Tafel斜率,表明Fe-aMOF具有最快的OER反应动力学.

表3 Fe基电催化剂析氧性能比较

图5 In-MOF、In1Fe1-aMOF、In1Fe3-aMOF、In1Fe4-aMOF、Fe-aMOF和NF的LSV曲线及其相应的Tafel图
为了进一步探究Fe-aMOF OER性能提高的可能因素,评估了催化剂的电化学活性表面积(ECSA).由于电极的ECSA与其双层电容(Cdl)成正比,因此可通过计算Cdl值的大小来反映催化剂的ECSA.图6(a)—图6(e)是In-MOF、In1Fe1-aMOF、In1Fe3-aMOF、In1Fe4-aMOF和Fe-aMOF在非法拉第区[电压范围0.1~0.15 V(vs.RHE)]、不同扫速下(20、40、60、80、100 mV/s)的CV曲线,在0.125 V(vs.RHE)处的ΔJ/2与扫描速率作图,得到直线的斜率即为Cdl[见图6(f)].Fe-aMOF的Cdl值为55.2 F/m2,高于In-MOF(29.5 F/m2)、In1Fe1-aMOF(34.1 F/m2)、In1Fe3-aMOF(47.2 F/m2)和In1Fe4-aMOF(50.7 F/m2)的Cdl值,表明Fe-aMOF的多孔性以及无定形结构赋予催化剂高的比表面积和更多裸露的活性位点,为电催化剂与电解质间提供有效的接触面积.

图6 In-MOF、In1Fe1-aMOF、In1Fe3-aMOF、In1Fe4-aMOF和Fe-aMOF在不同扫描速率时的CV曲线及在非法拉第区0.125 V( vs.RHE)处ΔJ/2相对于扫描速率的函数
利用电化学阻抗(EIS)探索电催化剂在OER过程中的电荷传输能力.图7为In-MOF、In1Fe1-aMOF、In1Fe3-aMOF、In1Fe4-aMOF和Fe-aMOF的Nyquist曲线,半圆的直径可以粗略反映电催化剂阻抗的大小,即阻抗由小到大的电催化剂依次为Fe-aMOF、In1Fe4-aMOF、In1Fe3-aMOF、In1Fe1-aMOF和In-MOF.通过等效电路模型拟合可得到界面转移电阻(Rct).其中:Fe-aMOF的Rct为1.04 Ω,远小于In-MOF的10.19 Ω、In1Fe1-aMOF的1.93 Ω、In1Fe3-aMOF的1.66 Ω和 In1Fe4-aMOF的1.58 Ω,说明Fe-aMOF与电解质界面处的电荷转移最快.此外,这些Rct值与电催化剂在电流密度为0.01 A/cm2时的过电位和Tafel斜率变化趋势相同,可见Rct是影响OER活性的主要因素.
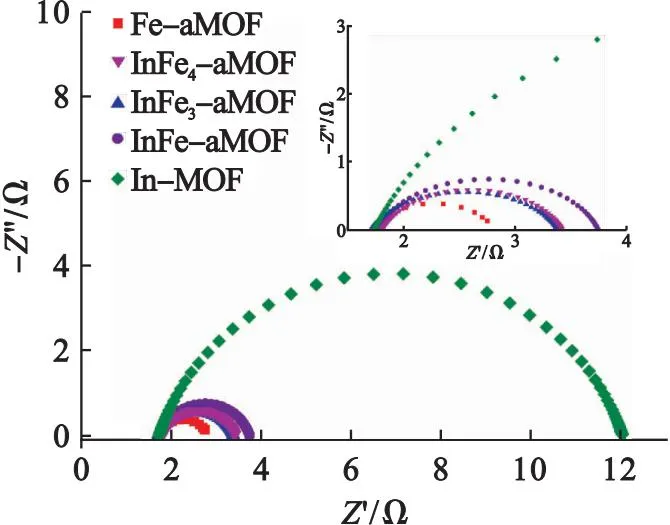
图7 In-MOF、In1Fe1-aMOF、In1Fe3-aMOF、In1Fe4-aMOF和Fe-aMOF的Nyquist图
除具有优异的OER活性外,电催化剂的稳定性也是评价能源转换系统的一个重要标准.如图8(a)所示,Fe-aMOF连续1 000圈循环伏安扫描后的LSV曲线与最初的LSV曲线非常相似,电流密度没有明显衰减.通过计时电流法评估Fe-aMOF的稳定性,结果如图8(b)所示.

图8 Fe-aMOF在进行1 000圈CV扫描前、后的LSV曲线和Fe-aMOF在0.01 A/cm2时的计时电流曲线
由图8(b)可以看出:连续测试40 h内,Fe-aMOF的电流密度几乎未出现较大波动,基本保持在0.01 A/cm2;连续测试超过40 h后,电流密度略微降低,可能是由电极表面产生大量气泡使催化剂从载体上脱落导致的.利用ICP分别对连续测试40 h和110 h后的电解液进行分析,未检测到金属离子(Fe3+和Ni2+),以上结果说明导电载体和Fe-aMOF催化剂在1 mol/L的KOH溶液中皆具有良好的化学稳定性.
3 结 论
通过简单溶剂热法合成了一系列无定形的InxFey-aMOF材料,并采用SEM、TEM、XRD、FT-IR和ICP等对样品的形貌、结构以及组成进行表征.研究结果表明:Fe3+的引入干扰了In3+与1,3,5-均苯三甲酸间配位,导致金属中心-有机配体配位缺失.这种外来金属离子诱导策略促使材料富有大量缺陷,暴露出更多的金属活性位点,从而提高OER电催化活性.电化学结果表明:在1 mol/L的KOH电解液中,Fe-aMOF表现出最佳的电催化活性:电流密度达到0.01 A/cm2时,过电位仅需要258.2 mV,塔菲尔斜率为45.4 mV/dec,优于目前报道的多数晶态MOF材料.该研究缺乏理论计算,后续研究应将实验结果与理论计算相结合,揭示催化剂结构与性能之间的构效关系.
