茸毛小长蝽的线粒体基因组测序和分析
2024-01-02林兴雨
林兴雨,宋 南
(河南农业大学 植物保护学院,河南 郑州 450046)
长蝽总科(Lygaeoidea)是半翅目(Hemiptera)蝽次目(Pentatomomorpha)中物种数量较为丰富的类群之一。目前,全世界有17个科超过4 290个物种已经被描述[1]。长蝽总科的大多数物种属于植食性昆虫,也有少部分是天敌昆虫。茸毛小长蝽(Nysiusgraminicola)隶属于长蝽总科长蝽科(Lygaeidae),是一些蔬菜和水果等作物上的重要害虫,尤其是在高粱、葡萄、番茄和桃等作物上危害较为严重[2]。
随着廉价且快速的二代测序技术的飞速发展,越来越多的昆虫高质量的线粒体基因组通过测序得到。昆虫线粒体基因组具有母系遗传以及进化速率较快等独特的优势,作为一种分子遗传标记被广泛应用于昆虫的系统发育研究中[3-5]。截至目前,NCBI数据库中已经有超过560条蝽次目的部分或完整的线粒体基因组序列被释放。近年来,利用分子学手段对蝽次目的高阶元的系统发育研究较多[6-7],而针对长蝽总科的内部的系统发育关系却研究较少。对长蝽总科系统发育研究的匮乏,不利于人们从分子学角度深入理解长蝽总科的系统发育关系。为了使用线粒体基因组数据更好地去了解长蝽总科的系统发育关系,本研究利用二代测序技术首次获得了茸毛小长蝽的线粒体全基因组序列,对其线粒体基因组序列进行了详细注释,分别分析了它的蛋白质编码基因、转运RNA基因、RNA基因,以及整个线粒体基因组的核苷酸组成、AT偏斜和GC偏斜、密码子使用频率情况、种间遗传距离,预测了茸毛小长蝽的转运RNA的二级结构。
此外,以GenBank数据库中已经释放的长蝽总科的17个科(象蝽科Idiostolidae、梭长蝽科Pachygronthidae、地长蝽科Rhyparochromidae、室翅长蝽科Heterogastridae、保长蝽科Artheneidae、尖长蝽科Oxycarenidae、皮蝽科Piesmatidae、Cryptorhamphidae、束蝽科Colobathristidae、Meschiidae、跷蝽科Berytidae、莎长蝽科Cymidae、Ninidae、杆长蝽科Blissidae、束长蝽科Malcidae、大眼长蝽科Geocoridae和长蝽科)52个物种的线粒体基因组序列作为内群,缘蝽总科Coreoidea的2个物种(Melanacanthusmarginatus和Notobitusmontanus)线粒体基因组序列作为外群,本研究基于线粒体基因组中的13个蛋白质编码基因的核苷酸序列矩阵和13个蛋白质编码基因的氨基酸序列矩阵,分别使用最大似然法(maximum likelihood,ML)和贝叶斯法(Bayesian inference,BI)重建长蝽总科内部的系统发育关系,明确茸毛小长蝽的系统发育位置,以期为更好地理解长蝽总科和茸毛小长蝽的系统发育关系提供线粒体基因组数据。
1 材料与方法
1.1 标本采集和DNA提取
本研究所使用的茸毛小长蝽的标本于2018年8月采集于河南省郑州市。将采集到的茸毛小长蝽标本浸泡在95%乙醇中,存放于河南农业大学植物保护学院标本馆并置于-20 ℃低温冰箱保存直到用于DNA提取。在实验室内使用天根公司的动物基因组DNA提取试剂盒(TIANamp Genomic DNA Kit,中国)按照说明书步骤提取茸毛小长蝽胸部肌肉的总DNA。使用NanoDrop 2000分光光度计对总DNA浓度检测以及配置1.5%的琼脂糖凝胶电泳进行总DNA的质量检测。
1.2 二代测序及线粒体基因组的组装
将检测质量合格且浓度大于50 ng/μL的高质量的DNA送往上海派森诺生物科技有限公司进行350 bp的小片段文库构建并进行二代测序,基于Illumina HiSeq 2500测序平台进行双末端(Paired-end,PE)测序。最后,将下机的原始数据去除接头,删除低质量序列,得到所需序列。利用GetOrganelle v1.7.5.2软件[8]对测序获得的clean reads进行线粒体基因组的组装和拼接,从而得到高质量的线粒体全基因组数据。
1.3 线粒体基因组、注释和分析
将组装得到的茸毛小长蝽的线粒体基因组数据在MITOS[9]网站上进行初步的基因注释,具体参数设置为:Genetic Code:05-inverterbrate;Reference:RefSeq 63 Metazoa,其余均按照MITOS的默认参数进行设置。通过MITOS预测获得茸毛小长蝽的22个转运RNA的二级结构。然后,通过近缘种线粒体基因组的序列比对和验证,手动调整蛋白质编码基因和RNA编码基因的基因边界。将注释完整的线粒体基因组序列上传至GenBank,得到茸毛小长蝽的GenBank登录号:OQ553818。利用系统发育分析学软件MEGA 7.0[10]分析茸毛小长蝽线粒体基因组的蛋白质编码基因、转运RNA基因和核糖体RNA基因的核苷酸组成以及蛋白质编码基因的密码子的使用频率情况。基于A、T含量之差与A、T含量之和的比计算AT偏斜,基于G、C含量之差与G、C含量之和的比计算GC偏斜。
1.4 多序列比对
采用MAFFT软件中的E-INS-I策略进行蛋白质编码基因的多序列比对[11]。使用TimAl v1.4程序对多序列比对中质量较差的序列进行剔除[12]。最后,运用FASconCAT-G_v1.04程序进行串联获得系统发育分析所用的数据矩阵[13]。用于系统发育分析的数据矩阵包括:(1) 13 个蛋白质编码基因的核苷酸序列构建的数据矩阵分别用于最大似然法和贝叶斯法系统发育分析; (2) 13 个蛋白质编码基因的氨基酸序列构建的数据矩阵分别用于最大似然法和贝叶斯法系统发育分析;
1.5 系统发育分析
本研究使用二代测序得到的茸毛小长蝽的线粒体基因组数据,并结合GenBank中获得的54种昆虫(52种长蝽总科昆虫作为内群,以及缘蝽总科2个物种作为外群)的线粒体基因组数据的蛋白质编码基因构建的2个数据矩阵。通过最大似然法(Maximum Likelihood, ML)和贝叶斯法(Mr Bayesian, BI)重建长蝽总科的系统发育关系。首先,使用 IQ-TREE 2.0.6[14]进行最大似然法分析:由软件自动筛选蛋白质编码基因的核苷酸序列矩阵最适模型为GTR+F+I+G4模型,蛋白质编码基因的氨基酸序列矩阵最适模型为mtZOA+F+I+G4模型,系统发育树的节点支持值通过自举检验置信度进行评估(运行次数设置10 000)。使用PhyloBayes v3.3 进行贝叶斯法分析[15]。首先,执行运算两次,每次运算包含四个链。核苷酸数据矩阵使用CAT-GTR模型以及氨基酸数据矩阵使用CAT-mtART位点异质模型来进行系统发育分析,利用PhyloBayes软件包中的bpcomp程序评估链间的趋同一致性。以最大差异值小于0.1作为分析达到有效的判定标准。最后,使用bpcomp程序构建50%合意树。
2 结果与分析
2.1 线粒体基因组分析
茸毛小长蝽的线粒体基因组结构图利用OGDRAW Version 1.1获得[16](图1)。茸毛小长蝽的线粒体全基因组长度为16 760 bp,其中A、T、G、C含量分别为43.10%、33.20%、9.50%和14.20%,其线粒体基因组包含37个基因和1个非编码控制区,H链(heavy strand)编码9个蛋白质编码基因和14个转运RNA基因(表1);L链(light strand)编码4个蛋白质编码基因和8个转运RNA基因以及2个核糖体RNA基因。此外,控制区位于rrnS和trnI之间,长度为2 011 bp,A、T、G、C含量分别为38.69%、36.20%、7.71%和17.40%。茸毛小长蝽的13个蛋白质编码基因、核糖体RNA和转运RNA以及整个线粒体基因组的AT含量为73.1%~78.3%(表2),均具有较高的AT含量,其中,位于L链的8个转运RNA基因的AT含量最低为73.1%,位于L链的4个蛋白质编码基因AT含量最高为78.3%。此外,AT偏斜和GC偏斜范围在-0.326~0.130和-0.198~0.291。

表1 茸毛小长蝽线粒体基因组注释Table 1 Annotation of the mitochondrial genome of N. graminicola
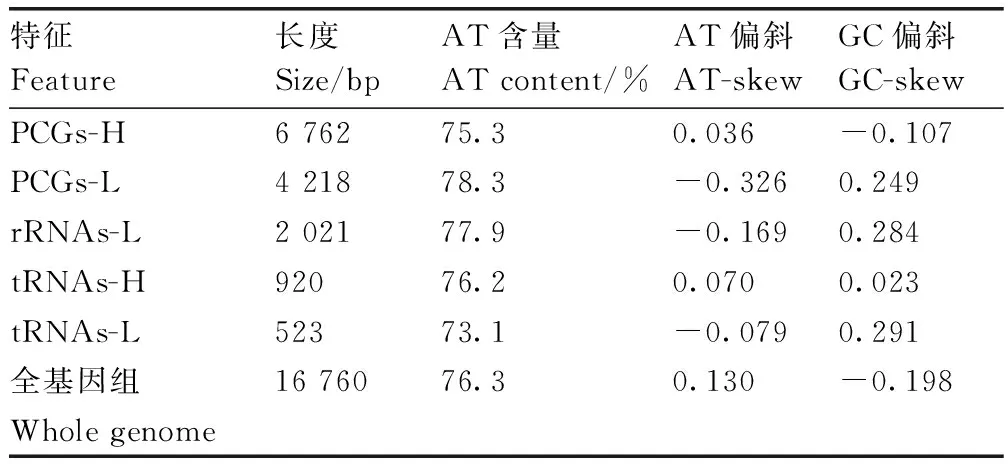
表2 茸毛小长蝽线粒体基因组的核苷酸组成和偏斜Table 2 Nucleotide composition and skewness of the N. graminicola mitochondrial genome
2.2 蛋白质编码基因分析
茸毛小长蝽线粒体基因组的13个蛋白质编码基因的序列在H链上的9个蛋白质基因序列总长为6 762 bp,AT含量为75.3%,GC含量为24.7%。L链上的4个蛋白质基因序列总长为4 218 bp,AT含量为78.3%,GC含量为21.7%。由上可知,位于H链和L链上的茸毛小长蝽的蛋白质编码基因均具有较高的AT含量。茸毛小长蝽的蛋白质编码基因的起始密码子除nad2利用GTG和cox1利用TTG作为起始,其余11个蛋白质编码基因的起始密码子都是以典型ATN密码子作为起始,除cox1、cox2、nad3、cob和nad1以不完整的T作为结尾,其余8个蛋白质编码基因的终止密码子都是以TAA作为终止密码子。在茸毛小长蝽线粒体基因组的密码子使用频率中,密码子AAA使用次数最多为427次,密码子GCG使用次数最少,仅使用2次(表3)。
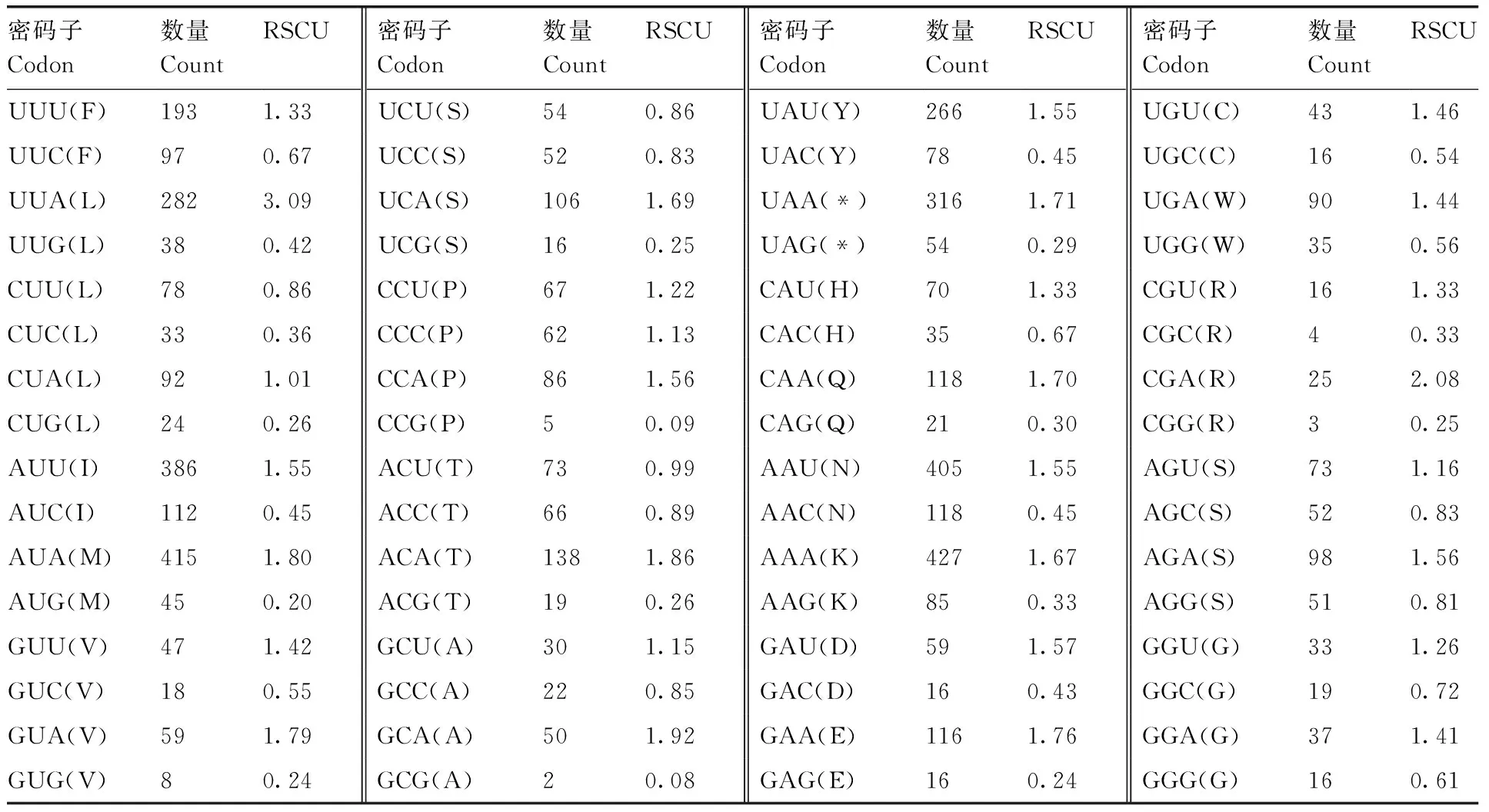
表3 茸毛小长蝽线粒体基因组的相对密码子使用频率Table 3 Relative synonymous codon usage of the mitochondrial genome of N. graminicola
2.3 tRNA和rRNA基因分析
茸毛小长蝽线粒体基因组的22个转运RNA基因的序列长度在61~73 bp范围内。其中trnA基因最短为61 bp,而trnK基因最长为73 bp。位于H链有16个转运RNA基因,序列全长为2 021 bp。AT含量为77.9%,GC含量为22.1%。位于L链6个转运RNA基因,序列全长为523 bp,AT含量为73.1%,GC含量为26.9%。只有trnS1由于缺少二氢尿嘧啶臂(DHU臂)从而形成一个简单的环,形成不完整的三叶草结构外,其余21个转运RNA基因都形成了完整的三叶草结构。此外,trnS1 的反密码子不是常见的GCU而是UCG(图2)。茸毛小长蝽的rrnL基因的序列全长为1 256 bp,AT含量为78.4%,GC含量为21.6%。rrnS基因的序列全长为765 bp,AT含量为77%,GC含量为23%。
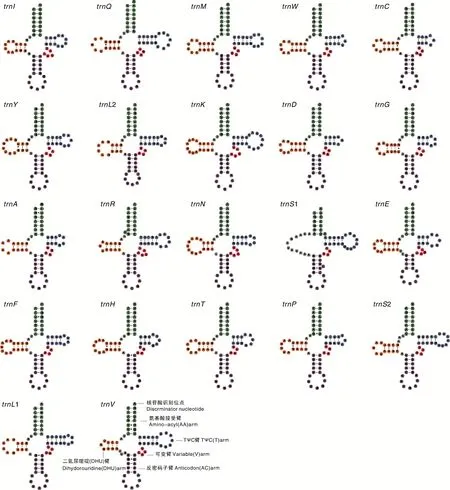
图2 茸毛小长蝽 22 个 tRNA基因的二级结构Fig.2 Secondary structure of 22 tRNAs genes of N. graminicola
2.4 系统发育分析
本研究结合长蝽总科的17个科的52个物种作为内群,缘蝽总科Coreoidea的2个物种作为外群,基于它们的蛋白质编码基因构建了核苷酸数据矩阵和氨基酸数据矩阵,分别使用最大似然法(ML)和贝叶斯法(BI)重建长蝽总科的系统发育关系(图3~图6)。系统发育分析结果一致显示:梭长蝽科、室翅长蝽科、跷蝽科、杆长蝽科和束长蝽科为单系群。地长蝽科和长蝽科为非单系群。然而,由于象蝽科、尖长蝽科、保长蝽科、皮蝽科、Cryptorhamphidae、Meschiidae、莎长蝽科、Ninidae、束蝽科和大眼长蝽科各仅有一个物种的线粒体基因组数据被释放,系统发育关系仍是不确定的。
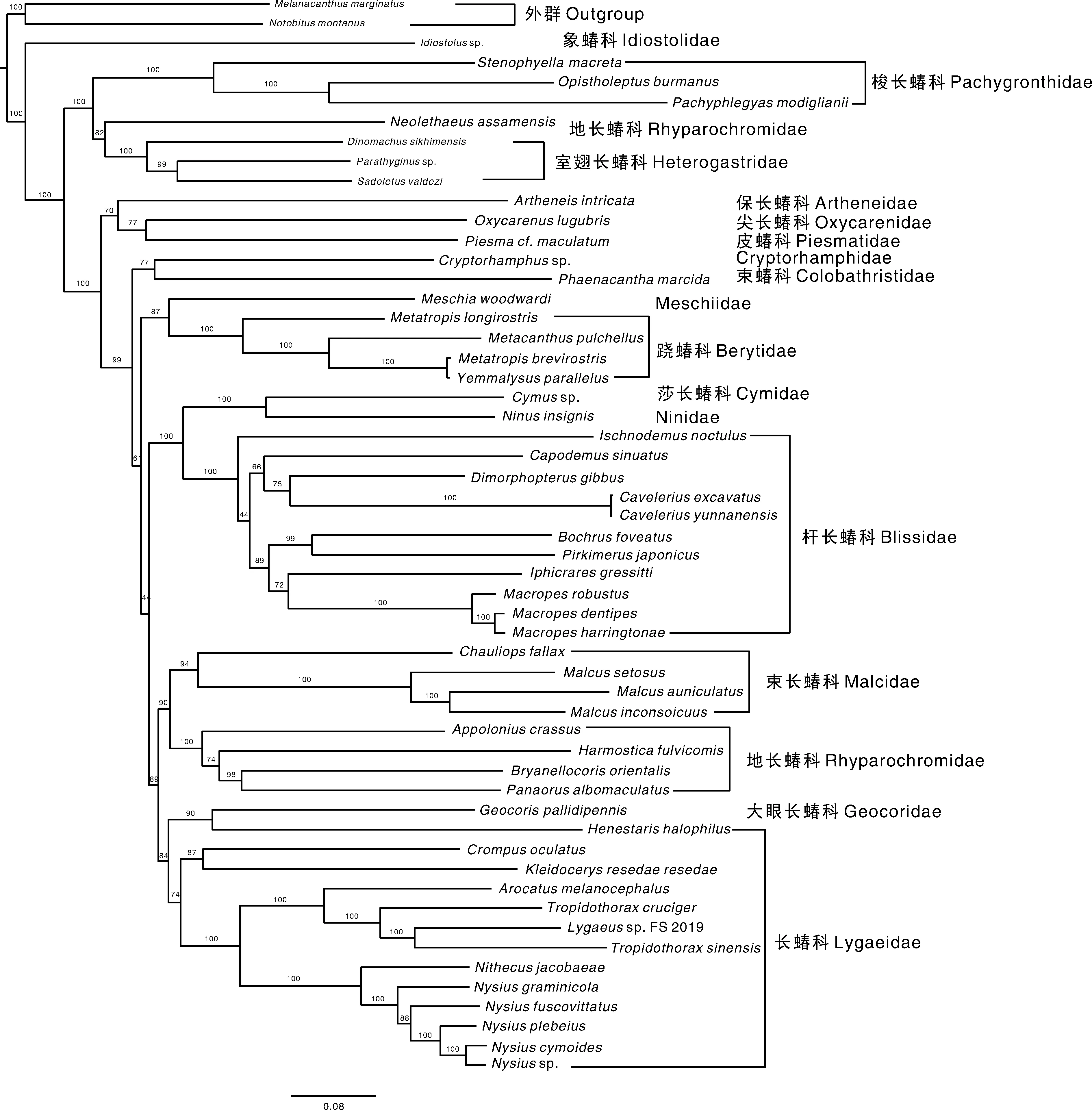
图3 基于线粒体基因组中13个蛋白编码基因核苷酸序列构建的最大似然树Fig.3 Maximum likelihood tree inferred from the nucleotide sequences of 13 protein-coding genes in the mitochondrial genome

图4 基于线粒体基因组中13个蛋白编码基因氨基酸序列构建的最大似然树Fig.4 Maximum likelihood tree inferred from the amino acid sequences of 13 protein-coding genes in the mitochondrial genome
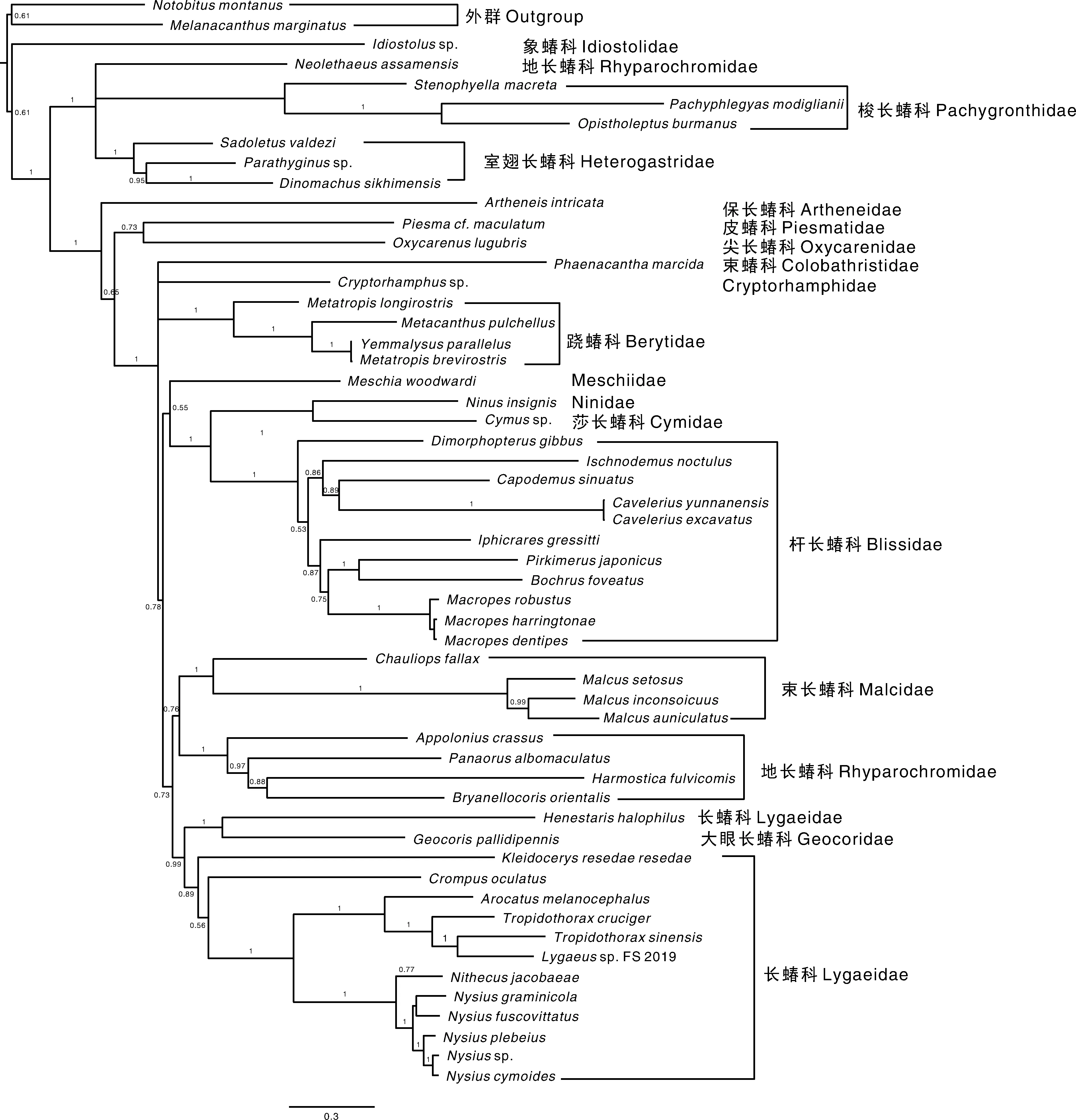
图5 基于线粒体基因组中13个蛋白编码基因核苷酸序列构建的贝叶斯树Fig.5 Bayesian tree inferred from the nucleotide sequences of 13 protein-coding genes the mitochondrial genome
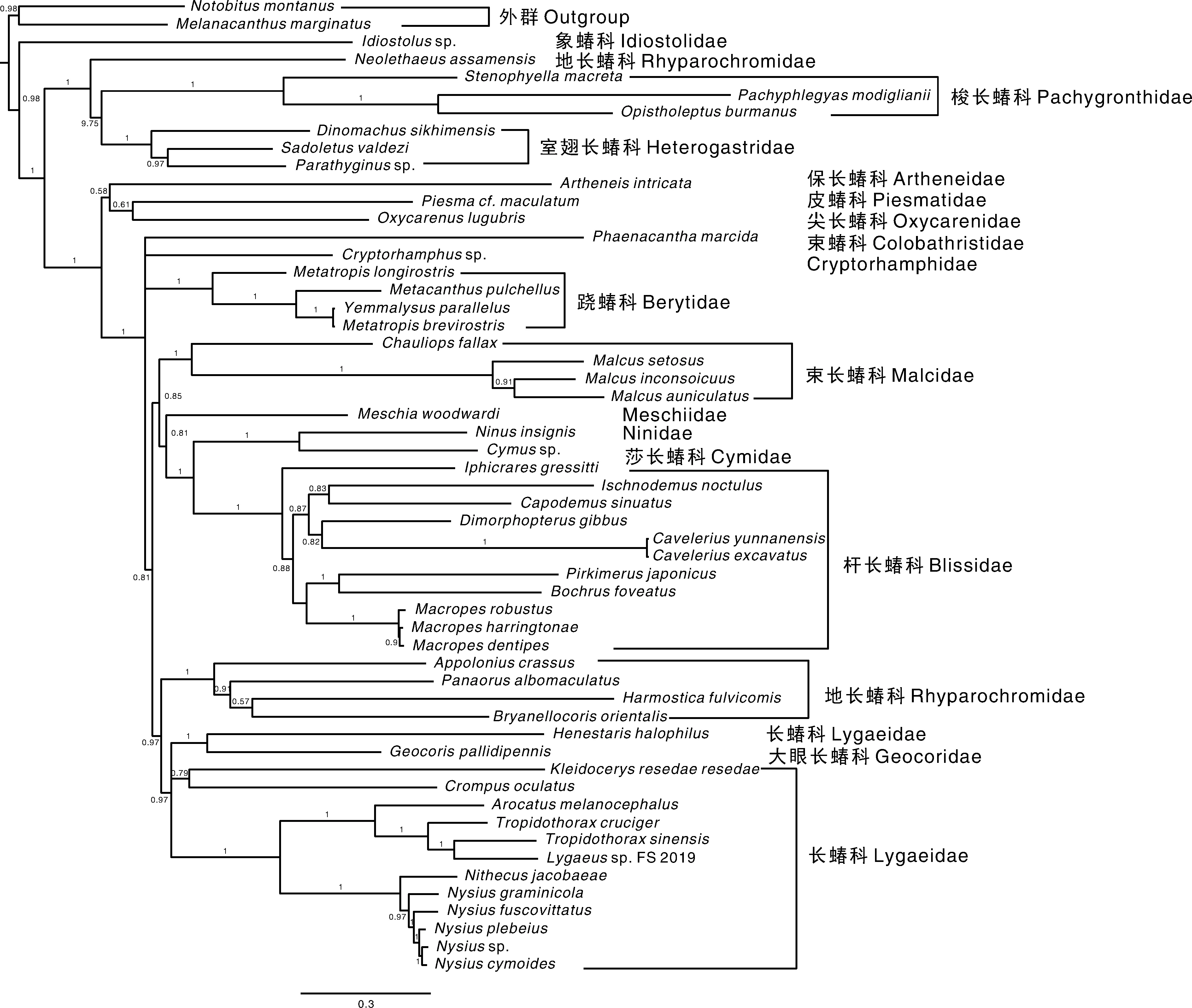
图6 基于线粒体基因组中13个蛋白编码基因氨基酸序列构建的贝叶斯树Fig.6 Bayesian tree inferred from the amino acid sequences of 13 protein-coding genes the mitochondrial genome
利用 Kimura-2-Parameter 模型计算了Nysius属5个物种(N.cymoides、N.fuscovittatus、茸毛小长蝽N.graminicola、N.plebeius和N.sp.)之间的种间遗传距离的结果显示:茸毛小长蝽与N.fuscovittatus在Nysius属的种间遗传距离最近为0.143。其次是茸毛小长蝽与N.cymoides和N.sp.的遗传距离为0.159和0.169,与N.plebeius的遗传距离结果最远为0.171(表4)。种间遗传距离结果与系统发育树分析的结果均支持茸毛小长蝽与N.fuscovittatus在Nysius属中具有相对较近的亲缘关系。
3 讨论与结论
本研究利用二代测序技术获得了茸毛小长蝽的线粒体基因组序列,分析了它的核苷酸组成等线粒体基因组结构,详细对茸毛小长蝽的线粒体基因组进行注释。茸毛小长蝽的线粒体基因组包含37个基因和一段非编码控制区。线粒体基因排列顺序与祖先昆虫的排列顺序一致,没有出现基因重排等特殊现象[3]。茸毛小长蝽的线粒体基因组的核苷酸组成与其它半翅目昆虫的核苷酸组成相同,都富含AT碱基[17]。此外,预测并绘制了茸毛小长蝽的22个转运RNA基因的二级结构。在所有转运RNA基因的二级结构中,只有trnS1基因缺少二氢尿嘧啶臂(DHU臂),其余转运RNA基因都具有完整的二级结构。这种情况在其它昆虫的转运RNA基因也出现过[18-24]。
在蝽次目的系统发育研究中,Li等[20]使用18S和cox1基因对蝽次目的系统发育关系进行初步研究的结果对长蝽总科的单系性是模糊的[7]。Yuan等[25]结合蝽次目的5个总科26个物种(其中涉及长蝽总科的5个科的6个物种)以及2个外群的线粒体基因组的蛋白质编码基因的核苷酸序列的系统发育结果显示长蝽总科的5个科(长蝽科、束长蝽科、跷蝽科、大眼长蝽科和束蝽科)为单系群[25]。此外,Huang等[26]使用长蝽总科的6个科(长蝽科、大眼长蝽科、跷蝽科、束长蝽科、地长蝽科和束蝽科)12个物种作内群以及2个物种做外群的蛋白质编码基因与核糖体基因利用最大似然法和Carapelli等[27]基于长蝽总科的6个科(长蝽科、束长蝽科、大眼长蝽科、地长蝽科、跷蝽科和束蝽科)13个物种作内群和2个物种作为外群的蛋白质编码基因利用贝叶斯法对长蝽总科内部的系统发育关系的研究结果一致显示地长蝽科为非单系群,其余5个科均为单系群。
本研究利用二代测序技术获得的茸毛小长蝽的线粒体基因组序列,并结合长蝽总科目前已经公布的所有线粒体基因组序列(包括17个科的52个物种)作为内群,2个缘蝽总科的物种作为群的线粒体基因组的蛋白质编码基因构建了蛋白质编码基因核苷酸序列矩阵和氨基酸数据矩阵,分别使用最大似然法和贝叶斯法进行系统发育分析,结果显示梭长蝽科、室翅长蝽科、跷蝽科、杆长蝽科和束长蝽科为单系群。地长蝽科为非单系群,这一结果与Huang等[26]和Carapelli等[27]的系统发育结果一致。然而,核苷酸数据矩阵利用最大似然法和贝叶斯法以及氨基酸数据矩阵利用贝叶斯法系统发育分析中由于大眼长蝽科的嵌入长蝽科而导致长蝽科为非单系群。此外,氨基酸数据矩阵利用最大似然法系统发育结果显示由于束蝽科的嵌入也导致了长蝽科为非单系群。
Nysius属5个物种的种间遗传距离与系统发育分析结果都显示茸毛小长蝽与N.fuscovittatus在Nysius属中具有较近的亲缘关系。这可能是由于它们在形态特征上以及自然界中的生物学习性上拥有较多的共同特点而形成的。
本研究利用二代测序技术首次获得了茸毛小长蝽的线粒体全基因组,分析了它的线粒体基因组结构特点,并基于数据库已经公布的所有的长蝽总科的线粒体基因组数据重建了目前较为全面的长蝽总科的系统发育关系。使用Kimura-2-Parameter 模型和两种系统发育分析方法明确了茸毛小长蝽的系统发育位置,为后续长蝽总科的系统发育研究以及茸毛小长蝽的种群遗传学等研究提供线粒体基因组数据。
