碘介导氧化型格氏反应制备α,β-不饱和酮
2023-12-26徐乔良安光辉李光明
徐乔良,张 聪,安光辉,李光明
(黑龙江大学 化学化工与材料学院,哈尔滨 150080)
0 引 言
α,β-不饱和酮类化合物广泛存在于天然产物和药物分子结构中[1-2],是复杂分子和天然产物合成中的重要中间体,也构成了各种重要反应的前体物,可通过亲电加成、亲核加成、迈克尔反应、Diels-Alder反应和插烯反应等过程来构建碳碳键[3-6]。同时,α,β-不饱和酮还成为近年来远端C-H活化官能化、不对称催化合成和脱氢芳构化的底物[7-10]。
经过多年的研究,开发了多种α,β-不饱和酮的合成方法。强氧化剂或者过渡金属催化氧化酮脱氢是制备α,β-不饱和酮的常用的方法之一。该方法使用当量强氧化剂(二氧化硒、DDQ等)来实现酮羰基邻位碳碳双键的构建,一般需要强氧化剂,且在高温下进行[11]。为了解决这一难题,又发展了羰基α位官能团化-消除的串联反应[12-15]。该方法通过合成α,β-不饱和酮。反应需要预先官能化,消除过程产生化学计量的副产物。近年来,电催化氧化脱氢,以及铱、钯、铂等贵金属催化也被开发来实现羰基化合物脱氢反应(图1)。

图1 金属催化脱氢反应制备α,β-不饱和酮Fig.1 Synthesis of α,β-unsaturated ketones by metal-catalyzed dehydrogenation reaction
除上述脱氢反应之外,一系列单电子氧化脱氢过程也被开发出来制备α,β-不饱和酮。Jung[15]课题组提出利用富电子的烯醇硅醚将电子转移到缺电子的三苯基正离子,实现烯醇硅醚氧化为不饱和羰基化合物的新思路(图2)。此外IBX[16]和AZADO[17]也被成功用于氧化酮脱氢制备α,β-不饱和酮。

图2 单电子转移氧化反应制备α,β-不饱和酮Fig.2 Synthesis of α,β-unsaturated ketones by single electron transfer oxidation reaction
除上述氧化还原过程,烯基和酰基亲电交叉偶联反应也被开发制备α,β-不饱和酮。使用酰基和烯基亲电试剂来制备α,β-不饱和酮被广泛研究,但试剂的可用性和成本效益仍有待探索(图3,path a和b)。为此,研究了过渡金属催化的亲电交叉偶联反应来制备α,β-不饱和酮[18](图3,path c)。该方法需要合成酰氟和三氟甲磺酸烯醇酯。需要注意的是一些酰氟不稳定,易于分解(图3)。
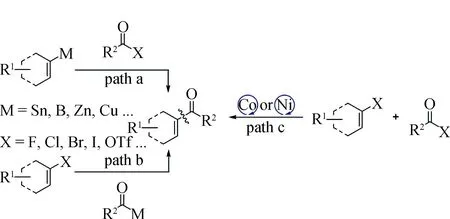
图3 亲电交叉偶联反应制备α,β-不饱和酮Fig.3 Synthesis of α,β-unsaturated ketones by cross-electrophile reaction
除上述方法外,通过亲核加成先制备醇,然后氧化得到α,β-不饱和酮。该方法第一步通过格氏试剂对α,β-不饱和醛亲核加成生成醇,或者醛和预官能化的烯烃通过Suzuki-Miyaura反应生成醇。第二步则通过Swern氧化生成α,β-不饱和酮。反应一般需要两步完成,且Suzuki-Miyaura反应需要当量的氯化铬,对环境有害[19](图4)。
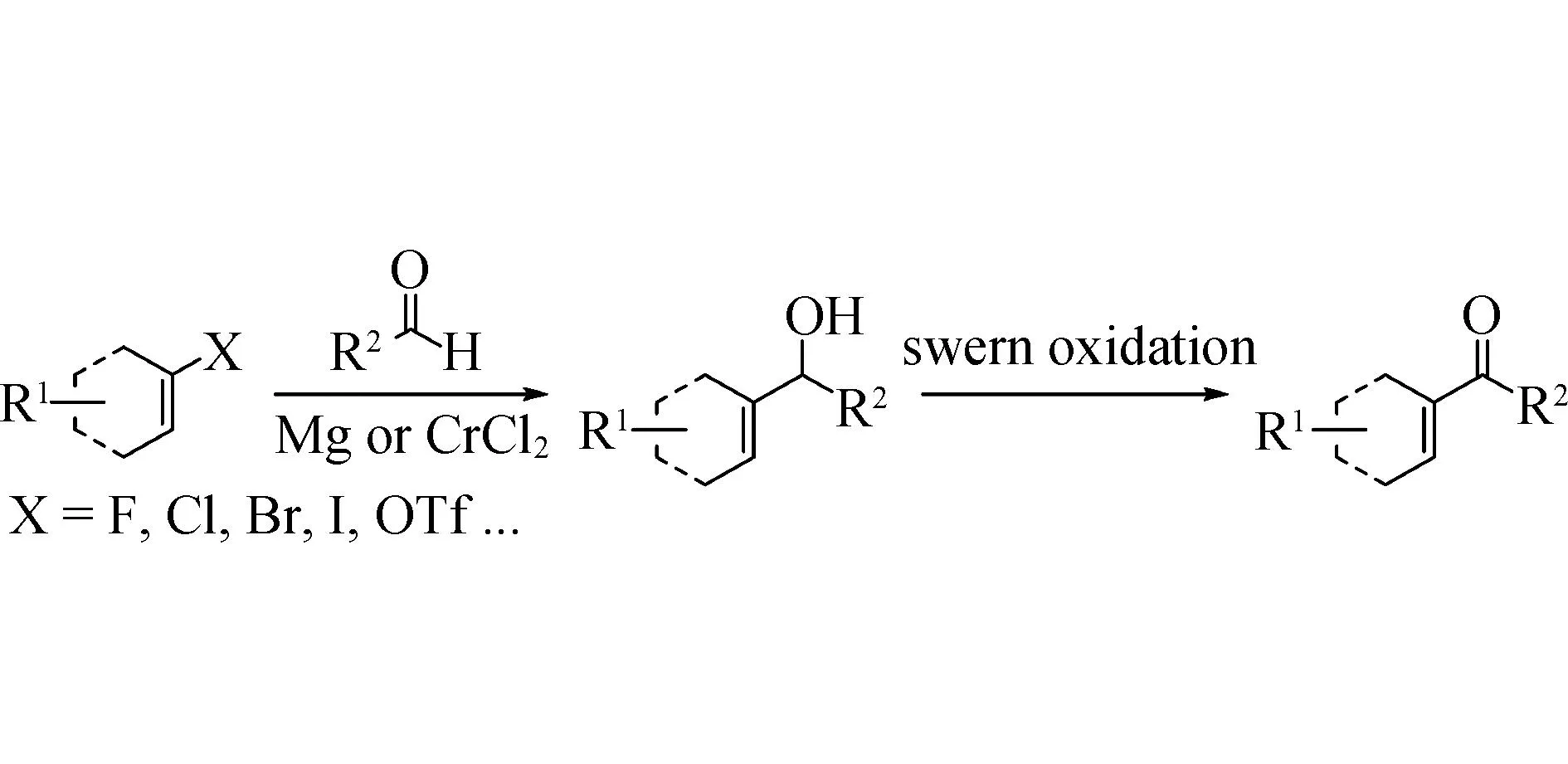
图4 亲核加成-氧化反应制备α,β-不饱和酮Fig.4 Synthesis of α,β-unsaturated ketones by nucleophilic addition-oxidation reaction
尽管很多方法可合成α,β-不饱和酮,但考虑到成本和操作难易,通过格氏试剂和不饱和醛的加成反应以及后续的氧化过程来实现α,β-不饱和酮化合物的合成具有一定的经济价值。该方法的问题在于需要两步合成[20-21]。为了解决该问题,提出一种碘介导格氏试剂和α,β-不饱和醛通过一锅反应生成α,β-不饱和酮。反应中碘既能引发格氏试剂的生成,又能将格氏试剂和α,β-不饱和醛进行1,2-加成反应生成的醇氧化为酮。实现高效合成α,β-不饱和酮化合物。
1 实验部分
1.1 实验试剂
实验所需的主要试剂见表1。

表1 主要使用的化学试剂名称及生产厂家
1.2 实验仪器
实验过程中使用的测试仪器名称、型号及生产厂家,见表2。
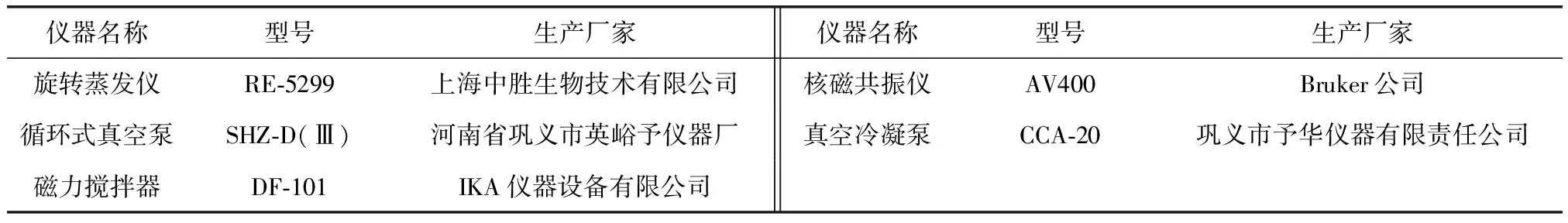
表2 实验仪器名称和型号
1.3 格氏试剂的制备
1.3.1 烷基溴化镁格氏试剂的制备
在氩气保护下,向烘干的史朗克管中加入镁粉(48 mg,2 mmol)、碘(254 mg,1 mmol)加入干燥的1 mL无水四氢呋喃。向上述混合物中滴加烷基溴(1 mmol)的无水四氢呋喃溶液(1 mL):滴加一点卤代烃的溶液,加热至回流引发,后持续滴入烷基溴,使其持续沸腾30 min,得到3种格氏试剂,密封待用(图5)。
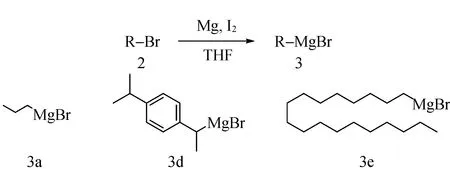
图5 烷基溴化镁格氏试剂的制备Fig.5 Preparation of alkyl magnesium bromide reagent
1.3.2 2-噻吩溴化镁格氏试剂的制备
在氩气保护下,向烘干的史朗克管中加入镁粉(48 mg,2 mmol)、碘(254 mg,1 mmol)和1 mL无水四氢呋喃。向上述混合物中滴加2-溴噻吩(1 mmol)的无水四氢呋喃(1 mL)溶液,加热至回流1 h,制备得2-噻吩溴化镁格氏试剂,密封待用(图6)。
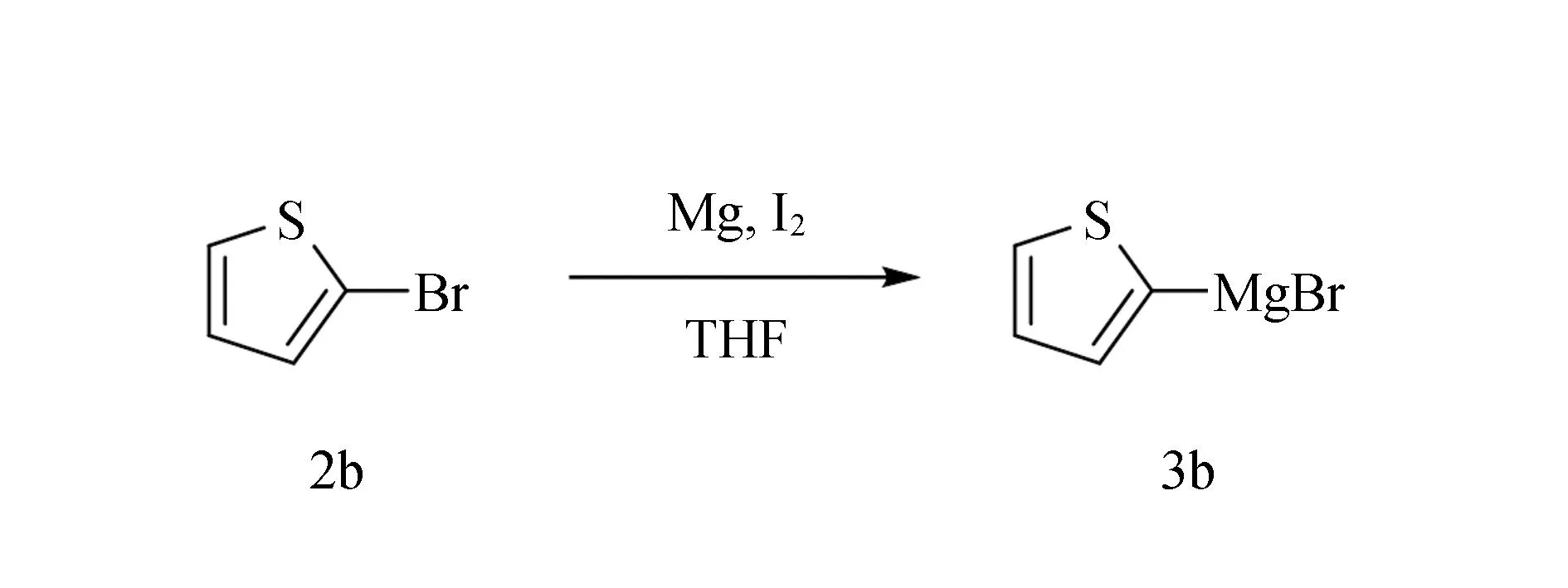
图6 2-噻吩溴化镁格氏试剂的制备Fig.6 Preparation of 2-thienylmagnesium bromide reagent
1.3.3 香茅基溴化镁格氏试剂的制备[21]
氩气条件下,将香茅醇(1.90 g,12.2 mmol)和吡啶(0.17 mL,2.1 mmol)加入无水乙醚(12 mL)中。将上述溶液冷却至-15 ℃,然后滴加三溴化磷(0.4 mL,4.1 mmol)。反应混合物在-15 ℃下搅拌2 h后,在室温下搅拌15 h。反应结束后,用无水乙醚(50 mL)稀释,将反应液倒入15 mL冰水中。用乙醚萃取(20 mL*2)。合并的有机相依次用饱和碳酸氢钠溶液(20 mL)和饱和氯化钠溶液(20 mL)洗涤。所得的有机相用无水硫酸镁干燥,过滤,减压除去溶剂。粗产品通过柱层析纯化(SiO2,石油醚)得到溴化香茅酯(1.07 g,40%)。在氩气保护下,向烘干的史朗克管中加入镁粉(48 mg,2.0 mmol)、碘(254 mg,1 mmol)和1 mL无水四氢呋喃。向上述混合物中滴加溴化香茅酯(219 mg,1 mmol)的无水四氢呋喃(1 mL)溶液:滴加少量卤代烃的溶液,加热至回流引发,后持续滴入溴化香茅酯溶液使其沸腾30 min,得到香茅基溴化镁格氏试剂,密封待用(图7)。
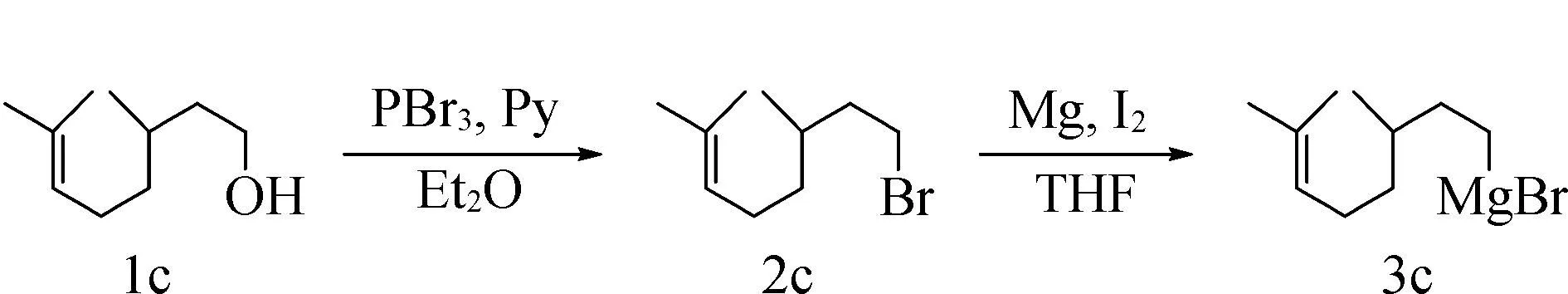
图7 香茅基溴化镁格氏试剂的制备Fig.7 Preparation of Citronella magnesium bromide reagent
1.4 α,β-不饱和酮的制备
在氩气保护下,向干燥的史朗克管中加入α,β-不饱和醛化合物(0.25 mmol)和无水四氢呋喃(1 mL)。在0 ℃下,将上述溶液滴加到制备好的0.5 mL(0.5 mmol·L-1)格氏试剂中,并缓慢升温至室温。反应在室温下搅拌12 h。反应混合物用饱和氯化铵溶液(20 mL)淬灭,之后用乙酸乙酯萃取(20 mL*3)。合并的有机相用无水硫酸钠干燥并减压浓缩。粗产品通过柱层析分离得到目标化合物(图8)。
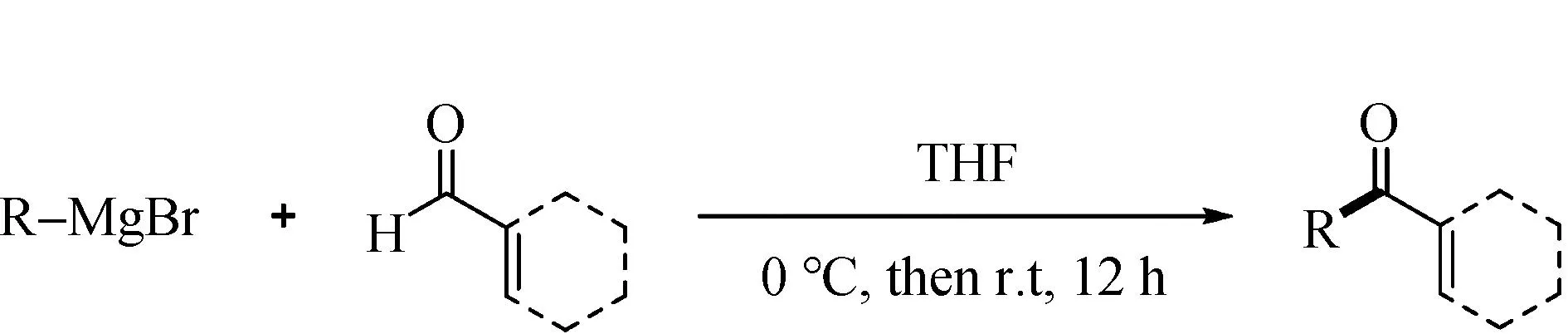
图8 α,β-不饱和酮化合物的制备Fig.8 Preparation of α,β-unsaturated ketones
2 实验结果与讨论
基于上述实验结果,不同格氏试剂的引发条件不同。格氏试剂的制备需要在无水条件下,格氏试剂引发的温度至关重要,长链烷基溴温度过低,不引发;温度过高,发生亲核取代反应。格氏试剂和α,β-不饱和醛化合物反应时,需要注意α,β-不饱和醛化合物滴加速度,防止局部温度过高导致副反应增多。
2.1 产率优化
探究了镁、碘的用量和溶剂对反应的影响。发现使用镁粉对α,β-不饱和酮的制备更有利,因为镁粉与反应接触面积大,有利于格氏试剂的制备,进而促进下一步反应。1 mmol的碘,既有利于格氏试剂的引发,又能把亲核加成生成的醇进一步氧化为不饱和酮。该反应溶剂必须进行严格除水,且溶剂沸点相对较高,筛选发现四氢呋喃作为溶剂,有利于α,β-不饱和酮的制备。
2.1.1 镁对反应的影响
由表3可见,镁的状态对产率影响很大。镁粉能更好地生成格氏试剂,反应效果最好。
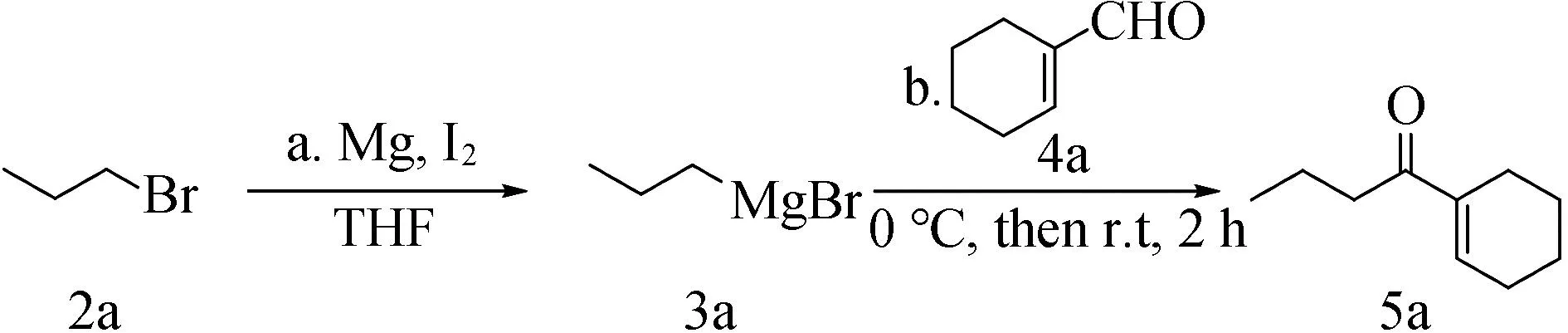
表3 镁对反应的影响
2.1.2 碘的用量对反应的影响
由表4可见,碘在制备α,β-不饱和酮化合物过程中,既是引发剂,又是氧化剂。不加碘,分离得到1,2-加成氧化生成1-(1-环己烯基)丁醇(5a﹡),产率为12%,不生成α,β-不饱和酮。额外的碘可用于氧化亲核加成之后的羟基。通过筛选碘的用量发现,1 mmol(254 mg)的碘,获得产率最高。当增加碘的用量时,产率无明显变化。
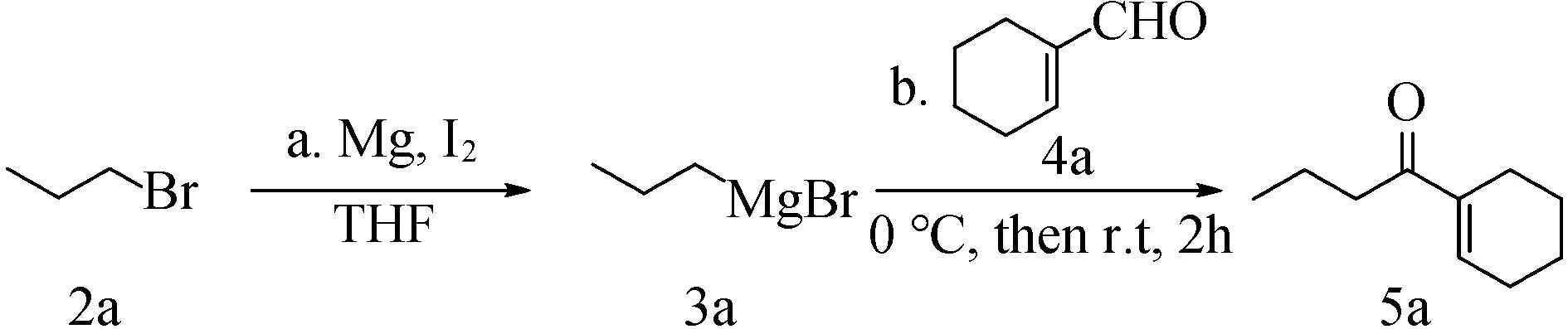
表4 碘的用量对反应的影响
2.1.3 溶剂对反应的影响
由表5可见,碘介导的α,β-不饱和酮化合物过程中,溶剂对反应有重要的影响。在烃类溶剂反应条件:a.在氩气条件下,2a(1.0 mmol)、Mg(2.0 mmol)、碘(x mmol)、2 mL四氢呋喃。b.在氩气条件下,3a(0.5 mL,0.5 mol·L-1)、4a(0.25 mmol)、1 mL四氢呋喃、0 ℃至室温搅拌2 h。
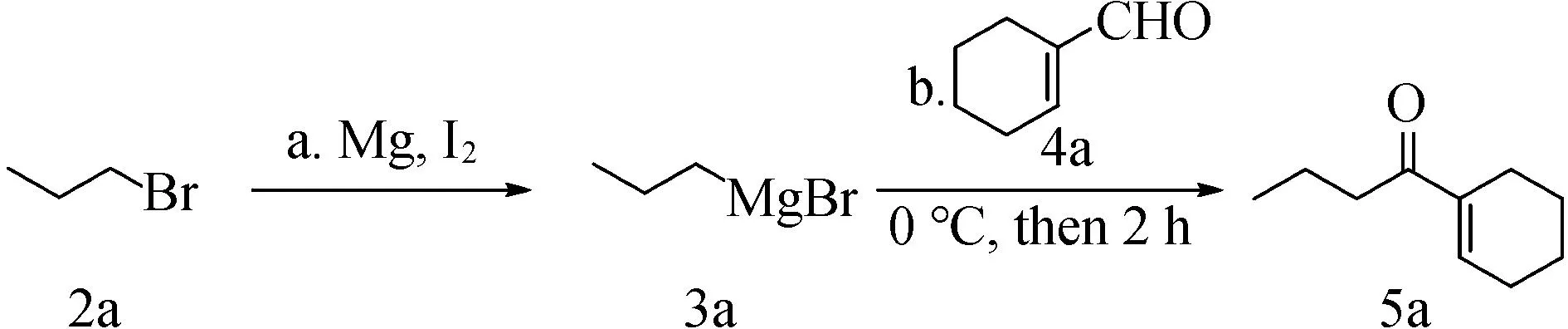
表5 溶剂对反应的影响
(如环己烷和甲苯)中,反应不发生。这主要是制备格氏试剂过程中,烃类溶剂不能稳定生成的格氏试剂。同为醚类溶剂,乙醚由于沸点低,不利于格氏试剂的生成,故也不能得到α,β-不饱和酮化合物。无水四氢呋喃既能稳定格氏试剂,又具有较合适的沸点,因而有利于格氏试剂的生成,以36%的产率生成相应的产品。
2.1.4 室温反应时间对反应的影响
由表6可见,格氏试剂和α,β-不饱和醛化合物室温反应过程中,延长反应时间对反应有重要影响。引发剩余的碘,有利于醇氧化生产α,β-不饱和酮,延长室温下反应时间,有利于氧化过程的进行。延长反应时间至12 h,得到最优产率57%。当反应延长至24 h,产率略微下降。

表6 室温下反应时间对反应的影响
2.2 底物拓展
在上述1.4实验条件下,制备得到8个α,β-不饱和酮化合物,并对其进行核磁表征。由5a~5e的产率对比可见,格氏试剂的空间位阻对反应产率有影响,空间位阻越小,产率越高。当选取空间位阻更大的(1R)-(-)桃金娘烯醛与十八烷基格氏试剂反应,以33%产率得到化合物5 g。由此可见,不饱和醛的位阻增大也降低反应的产率。除此之外,选取同一种格氏试剂分别和2-溴-1-环己烯-1-甲醛、肉桂醛反应时,不饱和醛的共轭体系增加,反应产率降低。由于共轭效应,当选择具有共轭体系的醛作为底物时,醛基不容易受到格氏试剂亲核进攻,使产品化合物5f、5h的产率相较5e降低(图9)。
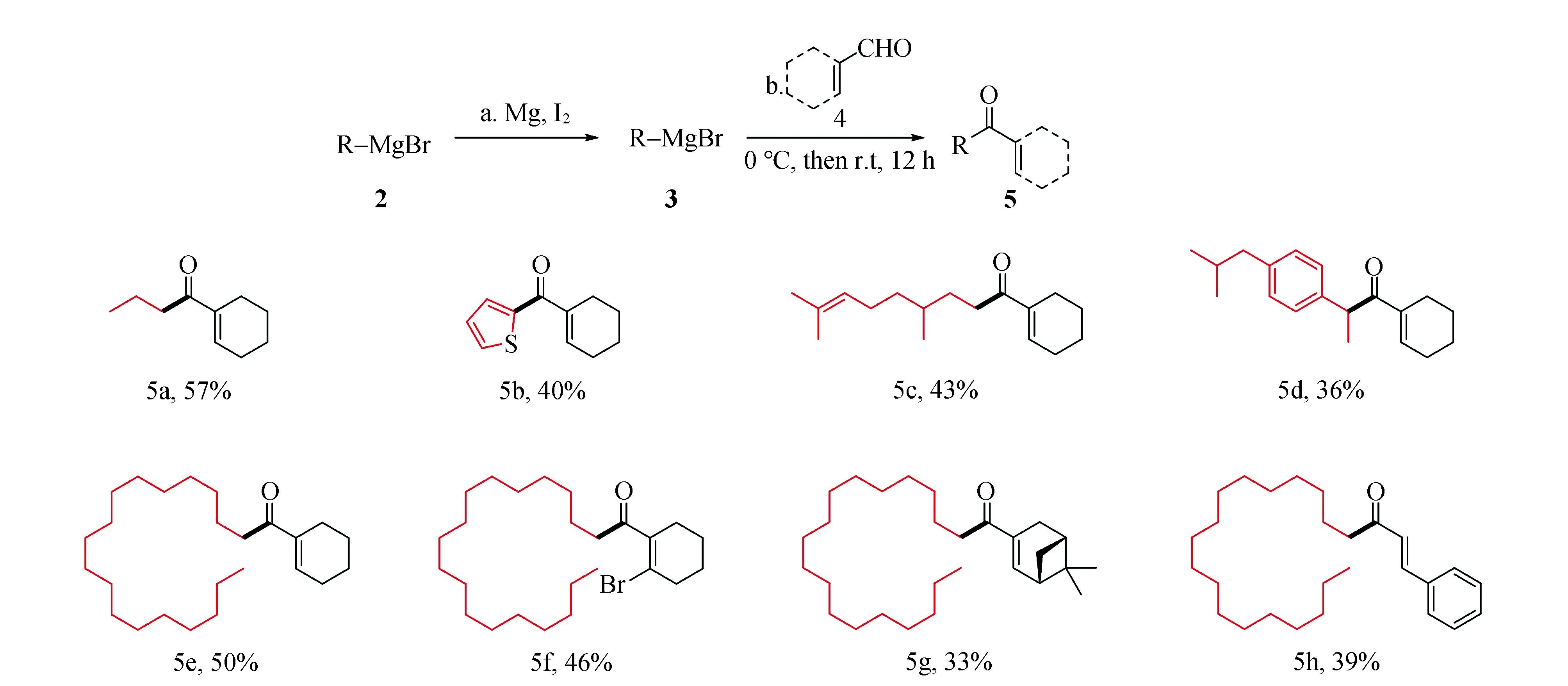
反应条件:a.在氩气条件下,2(1 mmol)、Mg(2.0 mmol)、碘(1 mmol)、2 mL溶剂。b.在氩气条件下,3(0.5 mL,0.5 mol·L-1)、4(0.25 mmol)、1 mL四氢呋喃、0 ℃至室温搅拌12 h。图9 制备α,β-不饱和酮化合物的底物范围Fig.9 Substrate range for preparing α,β-unsaturated ketones
2.3 产物的核磁表征数据
所合成的中间体和8种产物的核磁表征数据见图10。
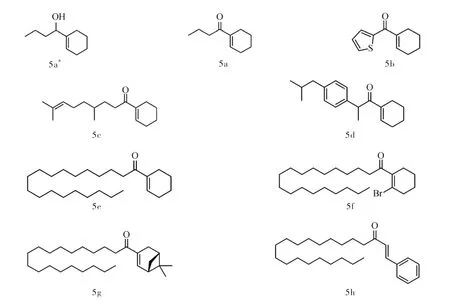
图10 所合成的中间体和8种产物的核磁表征数据Fig.10 HNMR of synthesized infermediates and eight products
化合物5a*由通用步骤获得无色油状化合物,产率12% (4.3 mg)。1H NMR (400 MHz,Chloroform-d)δ 5.51 (s,1H),4.46 (d,J = 5.0 Hz,1H),4.06~3.96 (m,1H),2.07~1.93 (m,4H),1.93~1.80 (m,2H),1.62~1.56 (m,4H),1.53~1.44 (m,2H),0.95 (s,3H)。
化合物5a由通用步骤获得无色油状化合物,产率57% (21.6 mg)。1H NMR (400 MHz,Chloroform-d)δ 6.85 (s,1H),2.58 (t,J=7.6 Hz,2H),2.27~2.13 (m,4H),1.65~1.53 (m,6H)0.86 (d,J=6.9 Hz,3H)。
化合物5b由通用步骤获得无色油状化合物,产率40% (19.2 mg)。1H NMR (400 MHz,Chloroform-d)δ 7.69~7.59(m,2H),7.35~7.26(m,1H),6.92~6.83 (s,1H)2.75~2.58 (m,2H),2.15 (t,J = 1.0 Hz,2H),1.78 (s,2H),1.60 (s,2H)。
化合物5c由通用步骤获得无色油状化合物,产率43% (26.9 mg)。1H NMR (400 MHz,DMSO-d6)δ 6.98 (s,1H),5.05 (t,J = 7.1 Hz,1H),2.67~2.53 (m,2H),2.19 (s,2H),2.07 (s,2H),1.96~1.87 (m,2H),1.62 (s,3H),1.57~1.45 (m,8H),1.34~1.23 (m,3H),1.11~1.03 (m,1H),0.82 (d,J = 6.4 Hz,3H)。
化合物5d由通用步骤获得无色油状化合物,产率43% (26.9 mg)。1H NMR (400 MHz,Chloroform-d)δ 7.11 (d,J = 2.4 Hz,4H),5.44 (s,1H),3.86 (q,J = 6.9 Hz,1H),2.97 (q,J = 14.1,13.4 Hz,2H),2.46 (d,J = 7.2 Hz,2H),2.00 (s,2H),1.92~1.79 (m,2H),1.76 (t,J = 4.6 Hz,1H),1.56~1.53 (m,2H),1.37 (d,J = 6.9 Hz,3H),0.91 (d,J = 6.6 Hz,6H)。
化合物5e由通用步骤获得白色固体化合物,产率50% (45.4 mg)。1H NMR (400 MHz,Chloroform-d)δ 6.89 (s,1H),2.60 (t,J = 7.6 Hz,2H),2.29~2.16 (m,4H),1.67~1.57 (m,6H),1.27~1.23 (m,30H),0.87 (d,J = 6.9 Hz,3H)。
化合物5f由通用步骤获得白色固体化合物,产率46% (50.8 mg)。1H NMR (400 MHz,Chloroform-d)2.73 (t,J = 6.0 Hz,2H),2.16 (d,J = 5.9 Hz,2H),1.71~1.64 (m,2H),1.67~1.57 (m,6H),1.27~1.23 (m,30H),0.87 (d,J = 6.9 Hz,3H)。
化合物5g由通用步骤获得白色固体化合物,产率33% (33.2 mg)。1H NMR (400 MHz,Chloroform-d)δ 6.75 (s,1H),2.95 (m,1H),2.25~2.11(m,4H),1.67~1.57 (m,4H),1.27~1.15 (m,31H),0.98~0.87 (m,9H)。
化合物5h由通用步骤获得黄色固体化合物,产率39% (37.5 mg)。1H NMR (400 MHz,Chloroform-d)δ 7.44 (d,J = 8.8 Hz,1H),7.24 (t,J = 7.8 Hz,1H),7.00~6.82 (m,3H),6.23~6.13 (m,1H),5.43 (q,J = 6.8 Hz,1H),1.82~1.61 (m,2H),1.34~1.24 (m,32H),0.90 (t,J = 6.7 Hz,3H)。
3 结 论
成功实现了碘介导下,格氏试剂和α,β-不饱和醛1,2-加成氧化制备α,β-不饱和酮。研究发现选用适量的碘以及沸点相对较高的无水四氢呋喃作为溶剂是实现反应的关键。当格氏试剂和α,β-不饱醛反应时,延长室温搅拌时间,有利于碘氧化醇生成α,β-不饱酮。该方法被成功用于格氏试剂与α,β-不饱和醛合成8个α,β-不饱和酮化合物。反应中碘既能引发格氏试剂的生成,又能将格氏试剂和α,β-不饱和醛进行1,2-加减反应生成的醇氧化为酮,合成化合物结构通过核磁共振氢谱进行了表征,将用于特殊合成方法的开发和天然产物转化研究。天然产物的研究与开发利用对于人类生态环境和经济发展都有着重要的意义。
