葛根药材HPLC 指纹图谱与6 种成分含量测定研究
2023-12-25闫小巧蔡鑫坤林敏生
叶 彬,闫小巧#,雷 婷,蔡鑫坤,林敏生*
1. 广州白云山中一药业有限公司,广东 广州 510530
2. 广州白云山奇星药业有限公司,广东 广州 510530
葛根药材为豆科植物野葛Puerarialobate(Willd.) Ohwi 的干燥根,习称野葛,具有解肌退热、透疹、生津止渴、升阳止泻等功效[1]。主要化学成分为3′-羟基葛根素、葛根素、3′-甲氧基葛根素、葛根素芹菜糖苷、大豆素、大豆苷等异黄酮类化合物[2-3]。在临床常用于治疗表证发热、麻疹不透、阴虚消渴、脾虚泄泻等症[4-5]。现代药理药效研究表明,葛根具有广泛的药理作用,如保肝、抗肿瘤、神经保护、心脏保护、改善胰岛素抵抗、抗炎、抗氧化等,在治疗心脑血管疾病、治疗糖尿病、抗肿瘤、预防骨质疏松、发挥雌激素作用等方面发挥重要作用[6]。
葛根目前已被国家卫生部批准列入“既是食品又是药品”名录,随着药食同源概念的普及与应用,葛根的市场开发潜力和使用需求量也日益增加。作为野生资源为主的中药材,有效保证其资源品质、控制其产品质量是葛根药材可持续利用的重要环节。因此亟需建立其质量控制和质量评价方法。据此,本研究建立葛根药材的高效液相色谱(HPLC)指纹图谱方法,该方法同时适用于葛根中6 种主要异黄酮成分的含量测定,并对从各主产区收集的样品进行了质量研究。以6 种葛根异黄酮总含量平均值及RSD、浸出物含量及RSD、相似度为指标,对不同产地葛根进行了质量比较研究,为制定优质葛根药材质量标准以及稳定原料产地提供方法和依据。
1 仪器与材料
Aglient HP 1100 高效液相色谱仪联用DAD 检测器(美国安捷伦科技公司)。对照品葛根素(批号200912)、染料木素(批号201312)、染料木苷(批号201702)、芒柄花素(批号201504)、大豆苷(批号201603)、大豆苷元(批号200402)、鹰嘴豆芽素A(批号200501)均购自中国食品药品检定研究院,质量分数均大于98%;对照品3′-羟基葛根素(批号CHB170224)、3′-甲氧基葛根素(批号CHB160902)、葛根素芹菜糖苷(批号CHB160421),质量分数均大于98%,均购自成都克洛玛生物科技有限公司。
从葛根主产区河南、陕西、四川、安徽等地收集样品共32 批,经鉴定符合《中国药典》2020 年版标准,为豆科植物野葛P.lobate(Willd.) Ohwi 的干燥根。经广州白云山中一药业有限公司陈自泓副主任中药师鉴定。样品信息见表1。
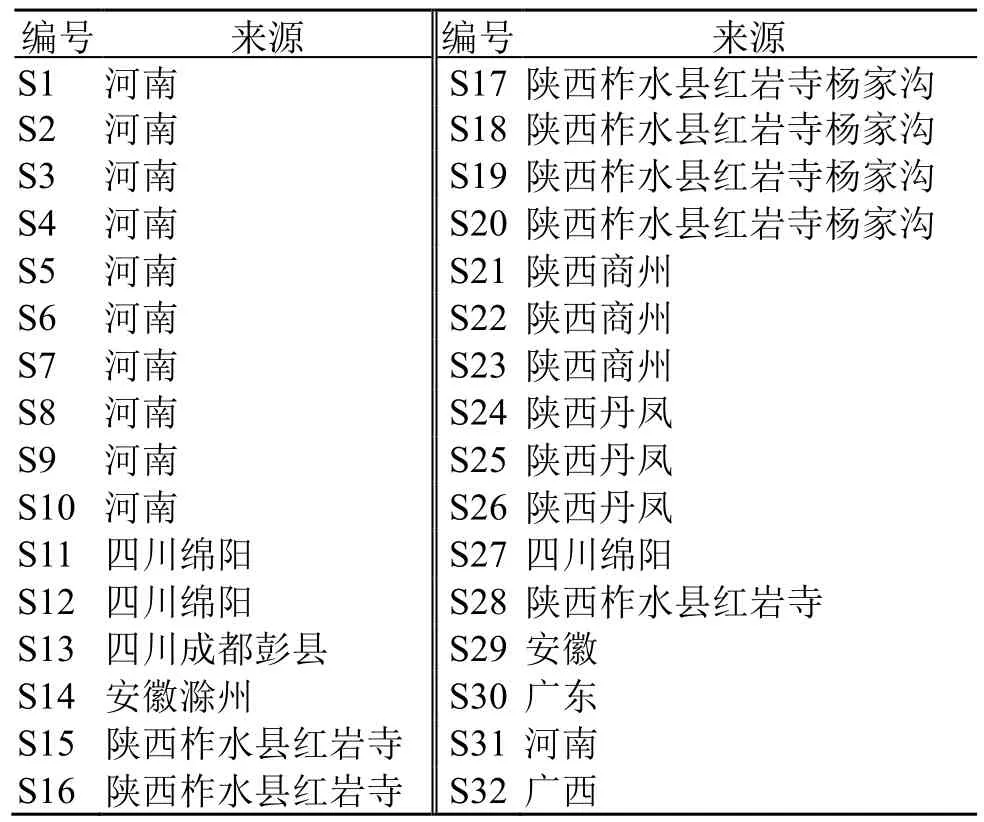
表1 32 批葛根药材样品信息Table 1 Sample information of 32 batches of Puerariae Lobatae Radix
2 方法与结果
2.1 指纹图谱的建立
2.1.1 色谱条件 色谱柱为Hypersil BDS C18柱(250 mm×4.6 mm,5 μm);流动相为0.1%甲酸水(A)-乙腈(B);梯度洗脱(0~2 min,5% B;2~3 min,5%~9% B; 3~20 min,9% B;20~21 min,9%~12% B;21~35 min,12% B;35~36 min,12%~15% B;36~50 min,15%~20% B;50~60 min,20% B;60~72 min,20%~50% B;72~80 min,50% B);体积流量为1.0 mL/min;检测波长为250 nm;柱温为25 ℃;进样量为10.0 μL。
2.1.2 供试品溶液的制备 精密称取葛根药材粉末0.1 g,置锥形瓶中,精密加入30%乙醇50 mL,称定质量,加热回流30 min,放冷,再称定质量,用30%乙醇补足减失的质量,滤过,取续滤液,用微孔滤膜(孔径为0.45 μm)滤过,滤液作为供试品。
2.1.3 对照品储备液的制备 分别精密称取约3′-羟基葛根素5 mg、葛根素10 mg、3′-甲氧基葛根素5 mg、葛根素芹菜糖苷5 mg、大豆苷5 mg、大豆苷元5 mg、染料木素0.2 mg、染料木苷4 mg、芒柄花素0.2 mg 定容至10 mL 量瓶,鹰嘴豆芽素A 0.1 mg 定容至20 mL量瓶,用甲醇溶解,制得对照品储备液。
2.1.4 对照品混合溶液的制备 精密吸取各对照品储备液3′-羟基葛根素5 mL、葛根素12.5 mL、3′-甲氧基葛根素5 mL、葛根素芹菜糖苷4.3 mL、大豆苷4.3 mL、大豆苷元2.4 mL、染料木素1 mL、染料木苷1 mL、芒柄花素1 mL、鹰嘴豆芽素A 1 mL,至50 mL 量瓶中,用甲醇制成含3′-羟基葛根素、葛根素、3′-甲氧基葛根素、葛根素芹菜糖苷、大豆苷、大豆苷元、染料木素、染料木苷、芒柄花素、鹰嘴豆芽素A 分别62.65、226.17、69.52、40.71、39.49、25.35、0.55、7.458、0.63、0.51 μg/mL 的混合对照品溶液。
2.1.5 精密度试验 取同一批葛根样品,按供试品溶液制备方法制备,取续滤液连续进样6 次,图谱导入相似度评价系统计算相似度,结果显示各图谱相似度均大于1,提示精密度良好。
2.1.6 重复性试验 取同一批葛根样品,照供试品溶液制备方法制备6 份,分别进样,记录各自指纹图谱色谱图,图谱导入相似度评价系统计算相似度,结果显示各图谱相似度均大于0.99。
2.1.7 稳定性试验 取同一批葛根样品,按供试品溶液制备方法制备,分别于0、3、6、9、12、24 h进样测定,图谱导入相似度评价系统计算相似度,结果显示各图谱相似度均大于0.99。
2.1.8 指纹图谱的建立 取32 批葛根样品,分别按照拟定供试品溶液制备方法制备,按拟定色谱条件进样分析,将所得色谱图导入相似度评价系统,以S1 为参照图谱,对照图谱生成方法为平均数,时间窗口1,经2、3、7、9 号色谱峰多点校正,Mark 峰匹配后,生成对照图谱(图1、2),并计算相似度,标记共有峰10 个,32 批样品与对照图谱的相似度均大于0.94。

图1 葛根样品HPLC 指纹图谱重叠图Fig. 1 HPLC fingerprint overlay of Puerariae Lobatae Radix
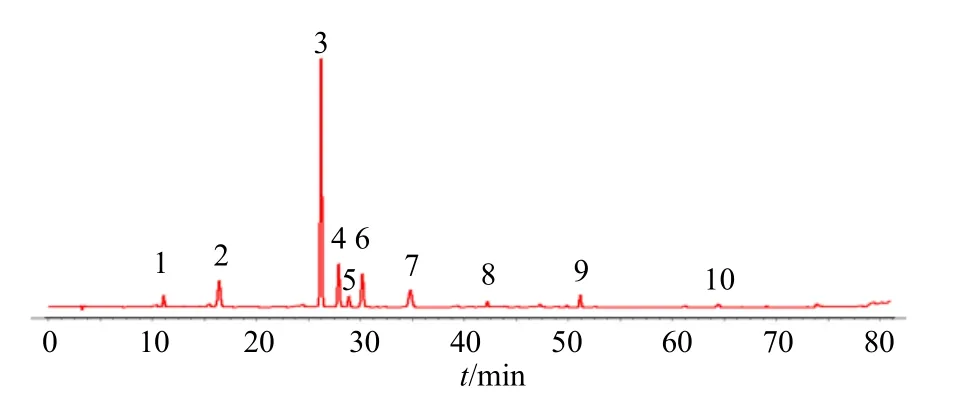
图2 葛根HPLC 对照指纹图谱Fig. 2 HPLC control fingerprint of Puerariae Lobatae Radix
2.1.9 指纹图谱共有峰的指认 通过对照品的保留时间,对葛根药材的指纹图谱共有峰进行指认(图3),初步确认了其中6 种化学成分,分别为1 号峰3′-羟基葛根素、2 号峰葛根素、3 号峰3′-甲氧基葛根素、4 号峰葛根素芹菜糖苷、5 号峰大豆苷、6 号峰大豆苷元。
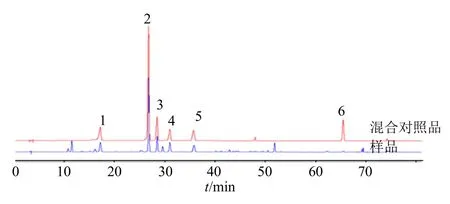
图3 葛根HPLC 指纹图谱色谱峰指认Fig. 3 HPLC fingerprint chromatographic peak identification of Puerariae Lobatae Radix
2.2 葛根HPLC 同时测定6 种异黄酮成分
2.2.1 色谱条件 同“2.1.1”项方法。
2.2.2 混合对照品溶液的制备 分别精密称取约3′-羟基葛根素2.5 mg、葛根素12.5 mg、3′-甲氧基葛根素5 mg、葛根素芹菜糖苷2.5 mg、大豆苷2.5 mg、大豆苷元1.25 mg,加甲醇溶解并定容至50 mL量瓶,制得含3′-羟基葛根素、葛根素、3′-甲氧基葛根素、葛根素芹菜糖苷、大豆苷、大豆苷元分别为55.02、254.72、101.74、53.76、53.02、27.48 μg/mL的混合对照品溶液。
2.2.3 供试品溶液的制备 同“2.1.2”项方法。
2.2.4 线性关系考察 精密吸取混合对照品溶液各1、2、4、6、8、10 μL,按色谱条件注入液相色谱仪,以峰面积为纵坐标(Y),以进样量为横坐标(X),绘制标准曲线。结果见表2。
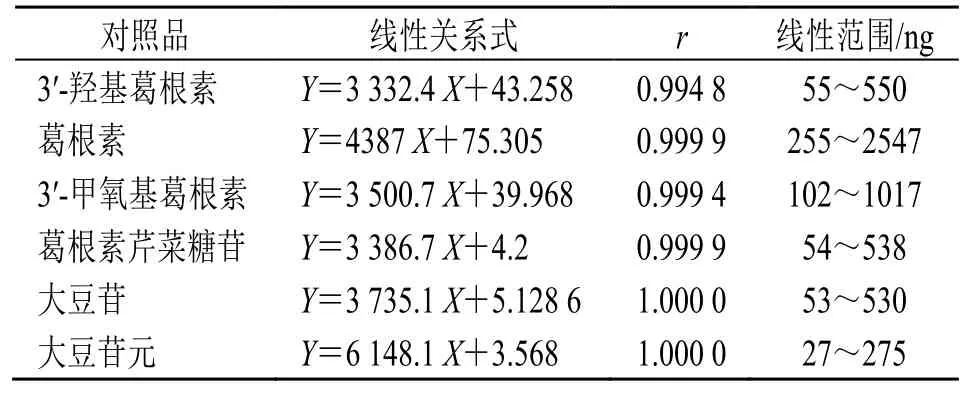
表2 对照品线性关系Table 2 Linear relation of reference substance
2.2.5 重复性考察 取样品(S1),平行6 份,精密称定,按上述方法平行制备成供试品溶液,分别进样10 μL 进行测定。结果3′-羟基葛根素、葛根素、3′-甲氧基葛根素、葛根素芹菜糖苷、大豆苷、大豆苷元质量分数RSD 分别为0.14%、0.16%、0.18%、0.18%、0.41%、3.95%。
2.2.6 精密度考察 精密吸取葛根样品(S1)的供试品溶液10 μL,连续进样6 次,测得3′-羟基葛根素、葛根素、3′-甲氧基葛根素、葛根素芹菜糖苷、大豆苷、大豆苷元峰面积的RSD 分别为0.10%、0.08%、0.17%、0.19%、0.41%、0.44%。
2.2.7 稳定性考察 取样品(S1)的供试品溶液,在室温下放置,分别在0、3、6、9、12、24 h 进样10 μL,测得3′-羟基葛根素、葛根素、3′-甲氧基葛根素、葛根素芹菜糖苷、大豆苷、大豆苷元峰面积的RSD 分别为0.31%、0.22%、0.28%、0.17%、0.49%、1.98%。
2.2.8 加样回收率考察 取已测定的样品6 份(粉碎,过40 目筛),每份0.05 g,精密称定,分别置锥形瓶中,精密加入3′-羟基葛根素、葛根素、3′-甲氧基葛根素、葛根素芹菜糖苷、大豆苷、大豆苷元的对照品储备液适量,按“2.1.2”项下方法制备供试液,进样测定,计算得到上述6 个成分的平均加样回收率分别为103.9%、104.7%、104.6%、96.8%、96.4%、96.4%;RSD 分别为0.94%、0.36%、0.80%、0.51%、1.44%、1.32%。
2.2.9 样品测定 按“2.2”项下方法制备供试品溶液,每批样品平行制备2 份,按“2.2.1”项色谱条件进样分析,并计算含量。结果见表3。
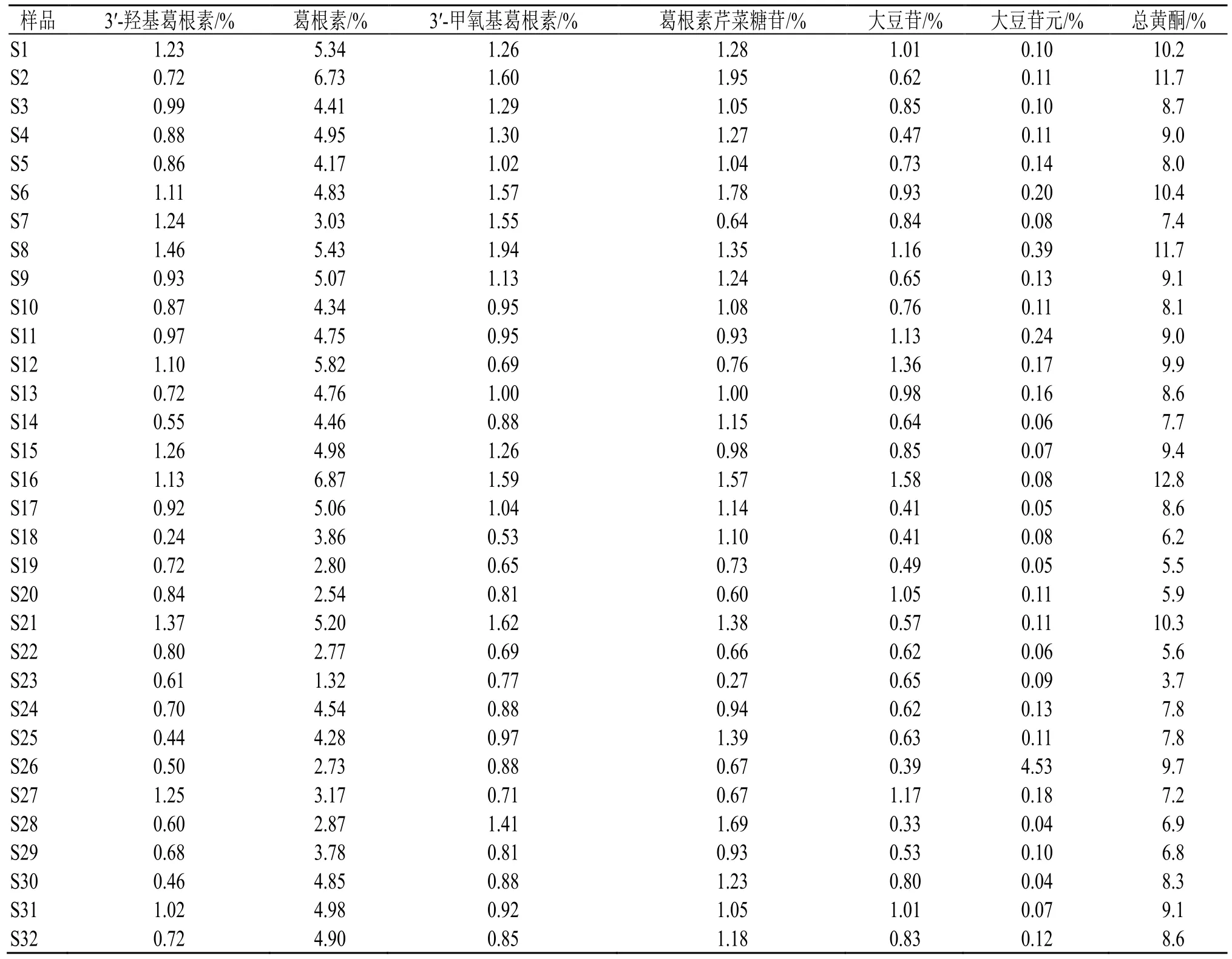
表3 32 批葛根多成分含量测定结果 (n=2)Table 3 Results of multicomponent content determination in 32 batches of Puerariae Lobatae Radix (n=2)
2.3 浸出物测定
按照《中国药典》2020 年版葛根浸出物项下方法对样品进行检测,结果见表4。
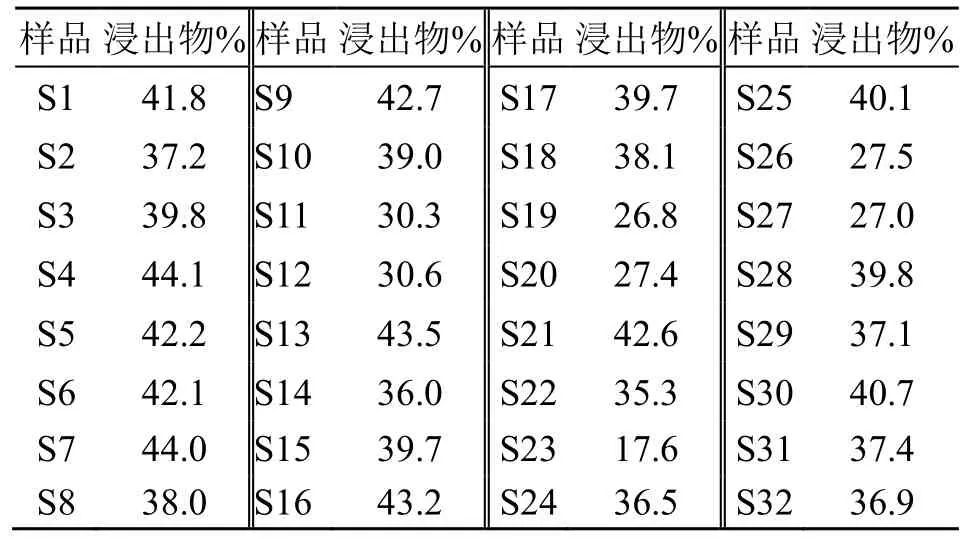
表4 32 批葛根样品浸出物测定结果Table 4 Determination of extracts from 32 batches of Puerariae Lobatae Radix samples
2.4 综合评分
通过考察,建立葛根HPLC 同时测定6 种异黄酮成分的方法。将不同产地葛根质量采用综合评分法对葛根药材质量进行综合评分(综合评分=黄酮总量×0.6+浸出物×0.4),结果显示(表5),河南产地的药材评分最高,其次为陕西。
3 讨论
3.1 供试品溶液的制备方法考察
本研究在初期考察了供试品溶液的制备方法,包括采用回流提取与超声的提取方法考察,结果从主要峰的积分面积可知,回流提取的效率高于超声提取,故选取回流作为提取方法。用不同体积分数(30%、50%、70%、100%)的乙醇作为提取溶剂,发现30%乙醇提取的成分色谱峰总积分面积最大,且30%乙醇提取的液相色谱图中各峰峰数较多,各主要成分色谱峰清晰可见,峰位峰形好,分离度高,故选取30%乙醇作为提取溶媒。对提取溶剂用量(15、25、50、75 mL 30%乙醇)考察发现,随着溶剂用量的增加,提取的样品主要成分含量越多,到50 mL 达到峰值后,含量水平趋于平稳,故选取50 mL 作为最佳溶剂用量。
3.2 色谱条件的比较与优化
对供试品溶液进行200~400 nm 的波长扫描,发现在250 nm 处可将全谱物质均检测出,各色谱峰的丰度、数目和分离度较好。同时还考察了不同色谱柱(Hypersil BDS C18、Hypersil ODS2、Spherisorb C18、Aglient Eelipee XDB-C18),发现Hypersil BDS C18柱分离效果较好。此外还对流动相(0.1%甲酸-乙腈、0.1%磷酸-乙腈、0.1%乙酸-乙腈、乙腈-水、甲醇-水)进行考察,结果发现采用0.1%甲酸水-乙腈流动相色谱峰分离效果好,峰数多、峰形好,且基线平稳。
3.3 分析与总结
《中国药典》2020 年版中对葛根水分、总灰分、醇浸出物测定作了规定,含量测定项下以葛根素的含量评价其质量。有学者认为以单一成分葛根素评价葛根的质量,存在一定的不科学性和不合理性[7]。中药指纹图谱可以较全面地反映中药所含化学成分的种类与数量,进而反映中药的质量和中医用药所体现的整体疗效[8]。目前已有研究关于采用HPLC 法建立的葛根药材指纹图谱研究,但所建立的指纹图谱中的化学成分不明确或研究成分数量较少[9-10]。本研究经色谱条件及方法学考察,通过测定32 批葛根药材,建立葛根药材的指纹图谱,提取出10 个共有峰,利用对照品保留时间共指认了其中6 个峰,分别为3′-羟基葛根素、葛根素、3′-甲氧基葛根素、葛根素芹菜糖苷、大豆苷、大豆苷元。相似度评价显示不同产地药材测定的指纹图谱与对照图谱相似度均大于0.94,所建立的指纹图谱稳定可靠,可用于葛根质量的评价。
采用葛根HPLC 同时测定6 种异黄酮成分,并对不同产区收集的32 份葛根样品进行质量研究。以3′-羟基葛根素、葛根素、3′-甲氧基葛根素、葛根素芹菜糖苷、大豆苷、大豆苷元6 种异黄酮成分的总和计算总黄酮含量,结合浸出物测定结果,综合评价不同产地的葛根药材质量。相比仅采用葛根素含量或少数成分含量作为不同产地葛根质量的评价指标[11-12],本研究针对葛根6 种异黄酮成分,并结合浸出物测定的综合评价方式更加全面,为制定优质葛根药材质量标准以及稳定原料产地提供参考。
利益冲突所有作者均声明不存在利益冲突
