基于不同π核扩展方式萘四甲酰基二酰亚胺有机半导体电荷传输性质的理论研究
2023-12-19潘佳峥孙晓琦任爱民郭景富
潘佳峥,孙晓琦,任爱民,郭景富
(1.东北师范大学物理学院,长春 130024;2.吉林大学化学学院,理论化学研究所,长春 130021)
近年来,随着科学技术生产力的不断提高,有机电子器件在生活中扮演着越来越重要的角色,如显示器、传感器以及银行卡上的电子条码等.这些应用推动着有机半导体材料(OSC)的迅速发展.OSC具有质量轻、价格低廉、可大面积加工、柔性大且可折叠等[1,2]无机半导体无法媲美的优点,因此,被广泛应用在有机场效应晶体管(OFETs)[3,4]、有机发光二极管(OLED)[5~7]、有机光伏电池(OPVC)[8]等电子器件中.OSC根据传输类型可以分为p-型传输、n-型传输和双极性传输3类.n型有机半导体是构建逻辑互补电路的重要组成部分[9,10],但在迁移率提高方面远远滞后于p型半导体.其在电子载流子传输的过程中阴离子极容易被环境中的氧气和水争夺电子而发生氧化反应,使得大多数n 型半导体在空气中稳定性差.Bao 等[11]总结n 型有机半导体材料的最低未占聚分子轨道(LUMO)能级应低于-3.15 eV,利于电子载流子的有效注入.
目前,实验已经报道了大量的n 型有机半导体材料.其中,包括萘四甲酰基二酰亚胺衍生物(NDI)、苝四甲酰基二酰亚胺衍生物(PDI)和寡聚对位苯乙烯基苯并二呋喃二酮衍生物(BDOPV)等,氯取代的NDI衍生物具有低的LUMO能级,其电子迁移率达到8.6 cm2·V-1·s-1[12].还有在PDI衍生物骨架的湾角处引入具有吸电子性质的取代基[如卤素原子、氰基(CN)],其中,PDIF-CN2是迄今为止空气稳定性最高的n 型有机半导体材料之一,通过实验测得的LUMO 能级为-4.5 eV,其电子迁移率达到6 cm2·V-1·s-1[13].而在空气环境条件下观察到的最高电子迁移率之一是四氟取代苯并二呋喃二酮,其电子迁移率高达12.66 cm2·V-1·s-1[14].NDI是一个常见的发色团且其衍生物具有良好的电荷传输性质,在NDI中心的核心位引入官能团可以自由调节其电化学性质和光学性质[15,16],Cl2-NDI的单晶场效应晶体管的电子迁移率是NDI衍生物中电子迁移率最大的,约为8.6 cm2·V-1·s-1[17].近年来,对NDI核心进行修饰或修饰酰胺取代基已成功获得p型、n型和双极性的低聚物或高聚物[18].Shukla 等[19]于2008年通过实验合成了NDI-C6(A1),其场效应晶体管的电子迁移率达到0.6 cm2·V-1·s-1.Yamada等[20]在2011年合成了TBI-C6(A2),实验测得其电子迁移率约为3.3×10-3cm2·V-1·s-1.2017 年,Zhang 等[21]合成了PyDI-C6(A3**),其电子迁移率达到了0.51 cm2·V-1·s-1.最近,Pei 等[22]合成了PyDI-C6(A3),并以A3分子为单元制成共轭聚合物PyDI-TT,其电子迁移率达到了0.335 cm2·V-1·s-1.2020 年,Madhu 等[23]合成了PDI-C6(A4),由于分子间的相互作用使A4分子拥有良好的光学性质,但其电荷传输性质暂时还未被报道.
由上述研究发现,以NDI 为核心骨架的π扩展的分子已被大量合成.但与p 型半导体[如二萘并[2,3-b:2',3'-f]噻吩并[3,2-b]噻吩(DNTT)通过在分子两端进行π扩展得到[2,3-b:2',3'-f]噻吩并[3,2-b]噻吩(DATT),空穴迁移率从8.3 cm2·V-1·s-1增加到16 cm2·V-1·s-1;并四苯扩展到并五苯、并六苯,空穴迁移率也逐渐增加]通过π扩展增加轨道间电子耦合相比,n型半导体情况比较特殊.常见的n型分子一般都含有吸电子基团以及卤素原子等,π扩展可能会打破分子的对称性.因此,对于n型材料π扩展对电荷传输性质影响的研究较少,尤其是沿不同方向的π扩展对n型半导体迁移率的影响还未被系统研究.本文基于几种典型的不同π核扩展的NDI有机半导体材料,利用第一性原理计算分析了其电荷传输性质,分别从单分子结构特征,分子间堆积方式以及分子间相互作用等方面阐明不同核心结构对载流子传输性质的影响.为了研究方便,定义垂直于酰亚胺取代基的方向为分子的短轴,而平行于酰亚胺取代基的方向为分子的长轴,基于A1分子,沿长短轴进行π扩展获得的系列研究分子的结构式见图1.
1 计算方法
有机半导体材料的电荷传输机制主要有3种模型:能带模型[24]、跳跃模型[25,26]以及介于两者之间的中间模型.能带模型要求晶体高度有序性,而跳跃模型适用于电荷局域在一个分子上的有机分子情形.采用跳跃模型基于半经典的Marcus理论进行电荷转移速率(kCT)[27]计算:
式中:ℏ为普朗克常数;kB为玻尔兹曼常数;T(K)为温度;λ(meV)为重组能;V(meV)为电荷转移积分;对于电荷自交换情况吉布斯自由能(ΔG0)为零.由于电荷传输是一种布朗运动,因此通过随机行走法来模拟晶体材料中的电荷扩散系数(D).基于X-ray晶体结构,对每个传输通道进行2000次随机行走模拟,每10 fs抽取一个样本,将这些样本的均方位移<x2(t)>对扩散时间t作图拟合得到D,使用课题组自编程序CTMP软件包[28]实现:
式中:n为传输维度;x(t)是电荷随时间t扩散的距离.
将D带入爱因斯坦公式[29]计算载流子迁移率(μ):
式中:e为电子电荷.由于晶体中分子堆积模式不同,因此电荷沿着不同方向传输速率不同,因此存在迁移率的各向异性,理论上各向异性迁移率(μ2D)的计算公式[30,31]如下:
式中:γi为层间传输方向与参考轴的夹角,当仅考虑二维平面传输时,γi为0;θi为第i个跳跃方向与参考轴夹角;Φ为目标方向相对于参考轴的方位角.
2 结果与讨论
2.1 分子的几何结构和电子结构
分子结构中烷基链和骨架间的二面角会受到环境影响,分别在气相、固相下优化了系列分子的几何结构.固相量子力学/分子力学(QM/MM)模型如图2所示,中心层分子采用 B3LYP/6-31G(d,p)水平,外层分子采用UFF力场[32],优化基于Gaussian 09程序包中的ONIOM模块[33,34].结果发现,固相下优化的几何更接近于晶体结构,表明晶体环境的极化对该体系影响较大.此外,可以发现沿A1分子短轴进行π扩展(A2)会使取代基与中心苯环之间的空间位阻增大,导致中心骨架发生扭曲(扭曲角可达13°~17°),而沿A1分子长轴进行π扩展(A3,A4)则不会增加酰胺基与中心苯环之间的空间位阻(表1).造成这一现象的主要原因是沿A1分子短轴π扩展,增加了氧原子与苯环外侧氢原子的空间位阻.
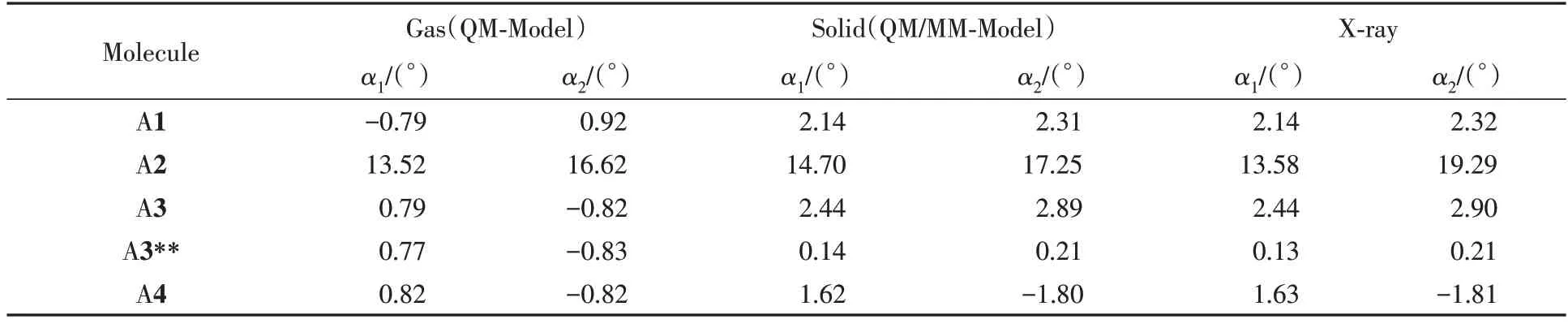
Table 1 Dihedral angles(α) between the acyl group and the central benzene ring of the series of molecules*
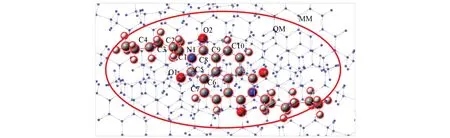
Fig.2 QM/MM model used to simulate the molecular geometries in crystal
载流子注入能力与前线分子轨道密切相关.为了探究沿A1长短轴进行π扩展后分子的空穴、电子的注入能力,在B3LYP/6-311+G(d,p)//B3LYP/6-31G(d,p)水平下计算了系列分子的轨道能级,结果示于图3和表2中.计算结果与实验结果接近,可以发现该系列分子的HOMO/LUMO均离域分布在整个骨架上,π扩展窄化了带隙(Eg),尤其是A2 分子的LUMO降低最大,HOMO相对提升.在实验中,金电极常被用于有机场效应晶体管(OFET)中,其功函为5.1 eV.与金电极相比,该系列研究分子的HOMO 能级均较低(-6.13~-7.31 eV),具有较高的空穴注入势垒,不利于空穴注入;LUMO能级的范围在-3.27~-4.04 eV 之间,电子注入势垒较低,有利于电子注入.因此系列研究的分子均具有成为n 型有机半导体材料的潜力,这与实验现象相符[26~29],因此,更加关注LUMO 能级的变化.相比于A1分子,A3和A3**分子的LUMO能级提高,这使得A3和A3**分子的电子注入能力未得到改善.为了找出长短轴扩展对LUMO能级影响的本质原因,选取不含杂原子的线性排列的萘(Naphthalene)、并四苯(Tetracene)以及聚集排列的芘(Pyrene)和苝(Perylene)进行对比,其前线分子轨道能级的结果列于表2.增加苯环会带来电子离域增加,LUMO能级均降低,其中苯环短轴线性扩展的Tetracene具有更低的LUMO能级.因此,A3分子的LUMO能级升高,不是因为苯环扩展带来的影响,而是酰亚胺基团和骨架共同作用所致.而A3与A3**分子除了骨架芘分子的取代位置不同,A3**分子的酰亚胺基团为五元环,两者能隙几乎不变,但A3**分子的HOMO/LUMO能级整体上移.这也说明酰胺取代基在芘分子的取代位置以及五元环的酰亚胺基团都会影响轨道能级.
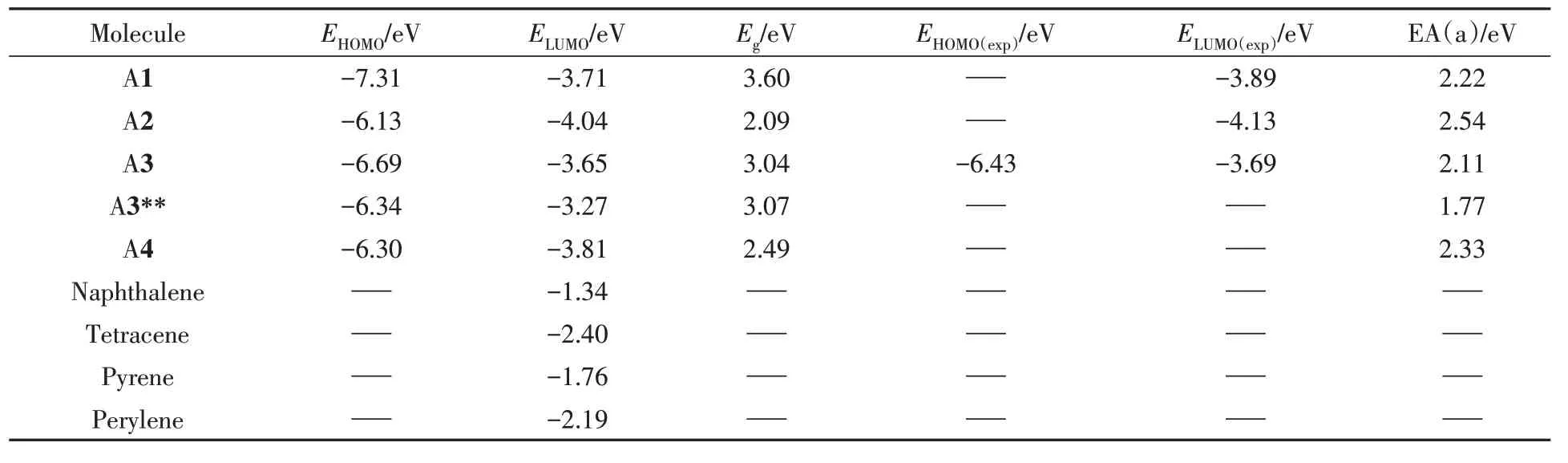
Table 2 Calculated electronic structures for the NDI derivatives and reference molecule at the level of B3LYP/6-311++G(d,p)//B3LYP/6-31G(d,p)
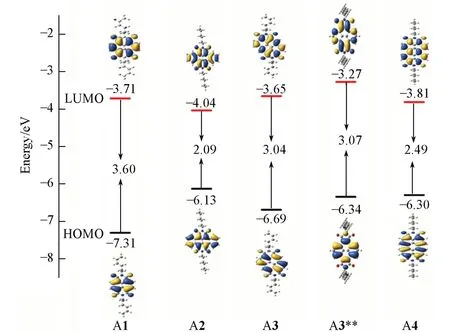
Fig.3 Distribution of HOMO and LUMO orbitals for the NDI derivatives
绝热电子亲和势(EA)的大小可表征分子抗氧化能力.计算了系列分子的平均绝热电子亲和势[EA(a)]值(表2).EA值越大,表明阴离子越易获得,越能有效地抵制外界环境中的氧气和水的氧化.所研究系列分子的EA的范围为2.11~2.54 eV,Chao等[35]和Ren等[36]研究表明,有机半导体材料的 EA数值接近或大于2.8 eV的分子可能具有在空气中稳定的电子传输.相比于A1,以A2方式π扩展后获得的EA值均得到了改进,为2.54 eV,比较接近空气中稳定的EA阈值;A1,A3以及A4分子的EA(a)值低于EA阈值,若想基于A1,A3以及A4分子制成空气稳定的n型OFET材料,则需要额外的保护措施,如贴防氧化膜以及放置在惰性气体中等.
2.2 重组能和正则模式
重组能(λ)是影响分子间电荷转移速率的一个重要参数.通过绝热势能面(AP)法和正则模式方法(NM)[33]分别计算了系列分子的电子重组能,结果列于表3.两种方法计算结果相近,表明系列分子在电荷传输过程中符合谐振子近似.其中,A1的电子重组能最大(346.4 meV),最小的是A3**(248.2 meV).结合前面的二面角几何结构和前线分子轨道分布可知,骨架的π扩展增加了分子几何的平面性的同时,带来了较大的轨道离域,进而降低了重组能.其中,平面性越好越有利于轨道离域,具有较好平面度的A3**分子在电荷转移过程中几何结构变化相对较小,因此其重组能也较小.而在引入苯环个数相同的情况下,沿短轴π扩展的A2的电子重组能要小于长轴π扩展的A3分子.
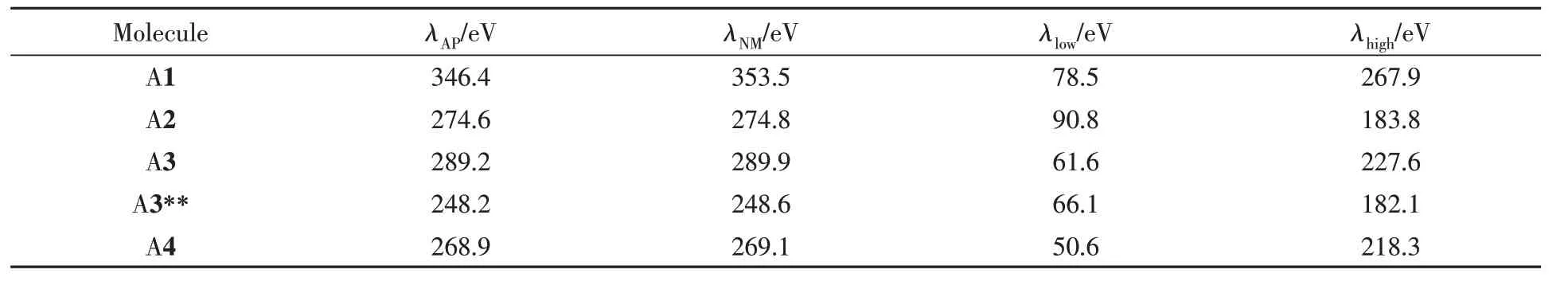
Table 3 Calculated total intramolecular electronic reorganization energies(λAP/λNM),and distribution in the high and low frequency regions(λlow/λhigh) with QM/MM model for studied NDI derivatives

Fig.4 Contribution of each vibration mode to the electronic reorganization energy(A1—E1)and the specific vibrational modes of the molecules corresponding to the vibrational frequencies(A2—E2) of A1(A),A2(B),A3(C),A4(D) and A3**(E)
为了深入了解长短轴扩展对重组能的影响,对电子重组能(λe)贡献较大的振动模式进行了分析(图4).振动频率低于700 cm-1的区域定义为低频区,其余为高频区,低频区和高频区对重组能的贡献列于表3,可以发现,不同的扩展方式分子在低频区产生的电子重组能变化相反,即沿长轴扩展的A3和A4分子低频区重组能降低,而沿短轴扩展的A2分子低频区重组能增加.为了说明这一特殊情况,研究了低频区的特殊振动模式(图4橙色框的区域),可见,A1分子在振动频率为456 cm-1处,振动模式为骨架面内碳原子的伸缩振动,产生的电子重组能大小为10.52 meV.A2分子在0~300 cm-1区域内由振动模式对应的电子重组能急剧增加,主要贡献源于在低频区沿分子短轴π扩展引入了额外振动模式(299 cm-1附近,表现为骨架面外摇摆振动,带来了较大的电子重组能,约15.6 meV).与A1分子相比,沿长轴扩展的A3与A4分子骨架的部分碳原子的伸缩振动以及氢原子摇摆振动被抑制,进而分子重组能降低.接下来,通过高频区振动模式对电子重组能的贡献分析发现,A1分子在高频区1471 cm-1附近对重组能贡献最大,高达46.04 meV,主要表现为中心苯环的碳原子的伸缩振动;然而π扩展后该频率下重组能急剧降低,说明沿不同方向π扩展均达到降低1471 cm-1处的振动幅度.通过上述分析可知,沿A1分子短轴进行π扩展得到的A2的重组能降低是由于高频区抑制作用导致的;而沿分子长轴进行π扩展的A3与A4,则在低频区和高频区均降低了电子重组能;此外,五元环的酰亚胺取代位置不同可以改变高频区的分子振动模式,相比于A3,虽然在低频区电子重组能增加,但是在高频区降低电子重组能的幅度更大,从而使A3**的电子重组能低于A3.
2.3 分子堆积、分子间转移积分和弱相互作用
转移积分是决定电荷转移速率的另一个关键因素,且其严重受到分子排列和取向的影响.系列分子的晶体堆积示于图5中.烷基链导致分子堆积的层间距离较大,电子耦合(转移积分,V)几乎为零,在此仅考虑层内分子堆积的转移积分.其中,A1分子表现为二维砖型π堆积,沿A1分子短轴进行π扩展形成A2分子,其晶体堆积方式转变为一维砖型π堆积,而长轴π扩展的A3和A3**分子也采取面对面的二维砖型π堆积,有趣的是,A4分子的堆积方式虽然为一维砖型π堆积,但其具有二维传输材料的性质.层内的电荷跳跃路径及转移积分分别定义为V1(红橙二体)、V2(红黄二体)和V3(红绿二体),其数值大小以及质心距离列于表4,是基于PW91/TZP 水平[37]利用格点能校正公式[28]计算获得的计算结果.其最有效传输通道为路径1 与路径2,表现为面-面堆积.A1 中路径1 的电子转移积分(VA1-e1=65.6 meV)较大,路径2和3的电子转移积分(VA1-e2=28.4 meV,VA1-e3=24.3 meV)要小很多;而基于A1分子短轴π扩展得到的A2分子,其路径1的电子转移积分急剧增加,高达91.8 meV,但是其余两条路径(VA2-e2=5.3 meV,VA2-e3=4.6 meV)非常小,属于一维传输材料,不利于材料的各向同性传输;而基于A1分子的长轴π扩展得到的A3(VA3-e1=65.1 meV),路径1的电子转移积分数值未增大,且路径2与路径3的电子转移积分数值变小;而A4 分子的路径1 的电子转移积分虽然也与A3 一样降低了(VA4-e1=33.0 meV),但其路径2的电子转移积分增加,因此,有利于电子的各向同性传输.
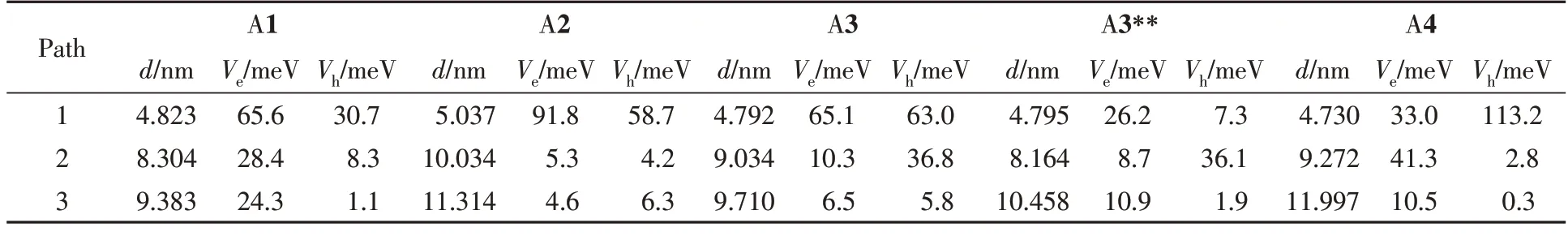
Table 4 Electron/hole transfer integrals(Ve/Vh) for the studied molecules,and the centre-of-mass distances(d) between neighbouring dimers
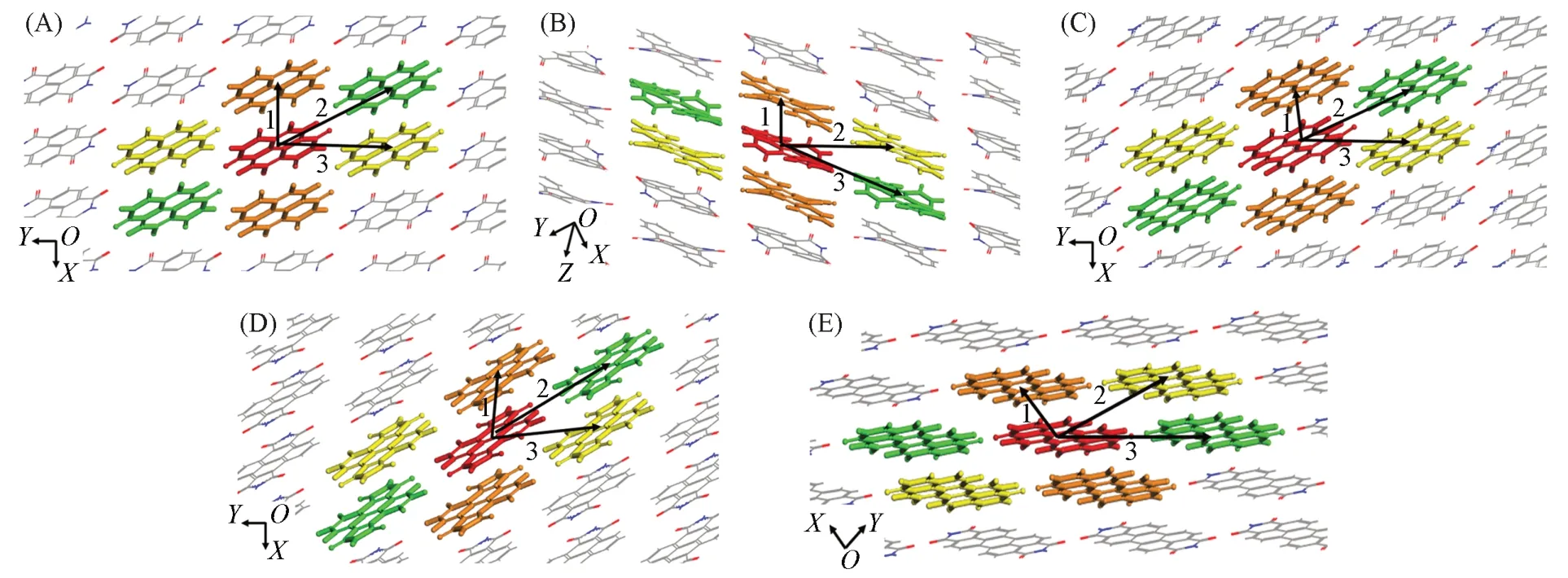
Fig.5 Crystal stacking patterns and major transport pathways of A1(A),A2(B),A3(C),A4(D) and A3**(E)
虽然A1~A4的主传输路径均是面对面堆积,但垂直距离、分子长短轴滑移距离均不同.需进一步分析长短轴滑移和垂直距离的改变对转移积分的影响,分子滑移示于图6中(黑色数字为分子长度;橙色为分子宽度;绿色为沿短轴的滑动距离;蓝色为沿长轴滑动距离;红色为面对面垂直距离).此外,计算了轨道重叠积分,进一步量化了每个分子电子轨道重叠程度(图7).可见,沿A1分子(34.1 nm)短轴进行π扩展的A2,其面对面二体短轴滑移距离(36.7 nm)增大,而A2沿长轴的滑移距离(8.4 nm)相比于A1分子(11.4 nm)却降低了,从侧面解释了A1分子短轴π扩展使其晶体的堆积方式从二维砖状堆积转变为一维π堆积的原因,也是导致A2电子转移积分(VA2-e1=91.8 meV)要高于A1(VA1-e1=65.6 meV)的原因;而沿A1 分子长轴π扩展得到的A3 与A4 分子,A3 在短轴滑移距离与A1 较为相近,面间距离略微增加,长轴的滑移明显减少,但这对电子转移积分影响不大.此外,从轨道重叠积分也可以看到,A1与A3轨道重叠接近(SA1-e=-0.0094,SA3-e=-0.0096),这主要是由于A1的S+较小,而S+与S-分别表示正负相位的轨道重叠积分,由于相位相反而抵消导致Se较小.相比于A1分子,A4分子长轴滑移距离急剧增加,短轴滑移距离明显降低,其路径1的电子转移积分(VA4-e1=33.0 meV)要远低于A1分子(VA1-e1=65.6 meV),而从轨道重叠积分图中也可看到,A4晶体中路径1的电子轨道重叠远小于A1分子.有趣的是,A4晶体的路径2的电子转移积分高达VA4-e2=41.3 meV,主要源于路径2的二体分子间垂直距离较小.综上所述,短轴进行π扩展,短轴滑移距离(36.7 nm)增大,长轴的滑移距离减小,表现为一维π堆积;长轴进行π扩展,长轴的滑移急剧增加或明显减少,短轴滑移距离减少或保持不变,因此堆积仍是二维π堆积(如A3),虽然A4显示为一维堆积,但其仍表现为二维电子传输性质,显然短轴扩展对堆积的影响更强.因此,结合轨道重叠和分子间相对位置,可以发现该系列分子的短轴滑移距离会影响轨道重叠,进而影响分子的传输维度,而分子长轴的滑移会影响面对面的二体的转移积分的大小.
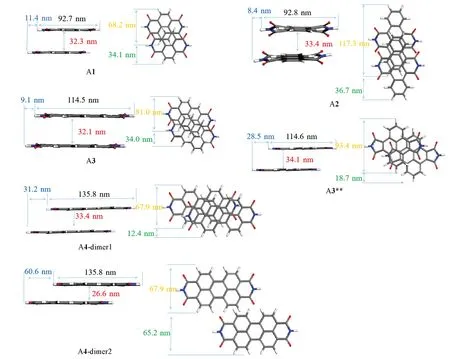
Fig.6 Displacements along the long-axis and short-axis for the face to face dimers extracted from the crystal structures

Fig.7 Orbital overlap integral for molecules
晶体分子堆积模式的形成源于分子间的弱相互作用.应用对称匹配微扰理论(SAPT能量分解[38])在SCS-SAPT0/jun-cc-pvdz水平上基于PSI4程序包[39]评估了所有分子的最近邻二聚体(传输路径1)的分子间相互作用能(ESCS-SAPT0)(表5).由于π扩展后分子组成和二体堆积结构都发生改变,使得绝对值之间的比较受限,因此,计算了每个子能量的百分比(表5中括号内的数值).相比于A1分子,引入π扩展会使交换排斥项的占比增加,而且A2的交换排斥能(Eexch)的占比以及数值大小均要略高于A4分子,且A2与A4分子面对面的二体的面面距离均为33.4 nm左右,将这种情况归因于A4分子的中心骨架趋近于平面化,分子的π共轭面积大导致交换排斥能增加.沿A1分子长轴π扩展得到的A3交换排斥能项占比也增大,但要低于A2分子.增大排斥力主要使A2和A3二体间的面面间距变大,A3分子的面面间距(32.1 nm)小于A2(33.4 nm);除此之外,沿A1分子短轴π扩展(A2)增加了静电相互作用能(Eelec)的占比,增幅为0.08%,相反,沿长轴π扩展(A3,A4,A3**)反而降低了静电相互作用能的占比,降幅为1.21%~4.23%.发现A1分子在引入π扩展后,吸引项[静电项、诱导项(Eind)与色散项(Edisp)之和]相比与排斥项占比更接近于2.0(除A3**),这也就意味着通过π扩展得到的系列分子的面对面分子对间的稳定性增强,此外,还计算了平均每原子的相互作用能为 1.63(A1),2.22(A2),1.92(A3),2.00(A3**)和2.22 kJ/mol(A4),再次验证了π扩展后得到的系列面对面二体分子间稳定性增强的结论.
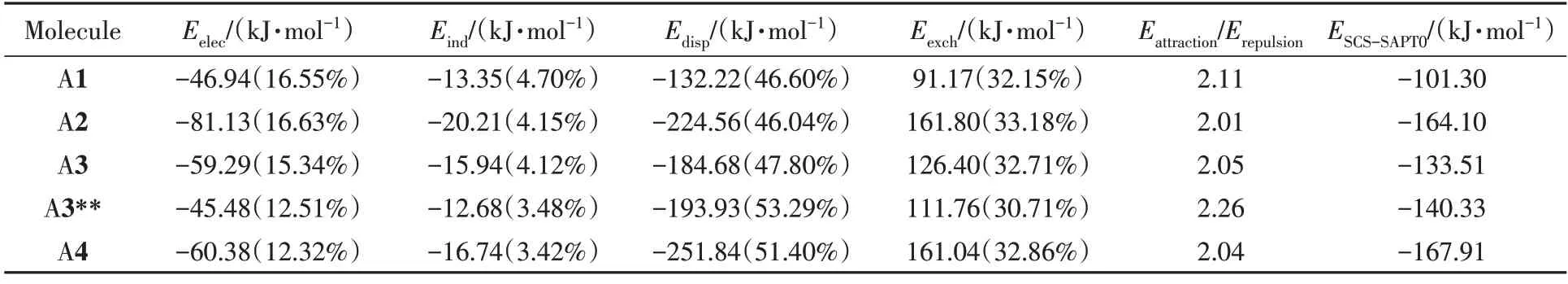
Table 5 Calculated intermolecular interaction energy of the nearest dimer for molecules A1—A4 and its corresponding energy decompositions by SAPT0
2.4 迁移率
迁移率是衡量电荷传输性质的重要指标,计算结果列于表6.其中,A1 路径1 的电子迁移率为0.19 cm2·V-1·s-1,基于A1 中心苯环沿不同方向π扩展得到的A2 和A3 路径1 的电子迁移率均明显增加,分别为0.96和0.32 cm2·V-1·s-1.有趣的是,A4分子最大传输路径的电子迁移率达到0.71 cm2·V-1·s-1,明显提高.虽然,A2分子沿着π…π堆积方向(路径1)的电子耦合较强,表现出较大的1D电子传输行为.但是在实验中,一维堆积的晶体材料的电荷传输可能受晶格缺陷影响较大.因此,开发具有大的二维平均电子迁移率、各向同性好的有机半导体实用性更广.理论评估二维平均迁移率的结果表明,该系列分子中A1分子的平均迁移率为0.07 cm2·V-1·s-1,短轴引入π扩展后,A2分子的平均电子迁移率降低(0.02 cm2·V-1·s-1),接近于实验测得的迁移率(0.03 cm2·V-1·s-1);长轴引入π扩展的A3分子的平均电子迁移率与A1相近;A4分子的平均电子迁移率最大,高达0.15 cm2·V-1·s-1.综上可知,沿A1分子中心苯环π扩展,短轴π扩展的A2在单维度传输具有明显优势;而长轴π扩展的A3与A4分子可能更具有二维传输材料的潜力.

Table 6 Simulated electron mobilities of all compounds investigated and available experimental values*
*μ1Dmax,μ2Drepresent the mobility along the most efficient hopping pathway and the average 2D hole mobility,respectively.
为了更好地了解分子晶体中不同方向电荷传输性质,计算了研究分子的二维各向异性迁移率(图8).其中,A1的各向同性迁移率最优,在Φ=30°时,具有最大的电子迁移率约为0.488 cm2·V-1·s-1,与实验测得的最大电子迁移率(0.70 cm2·V-1·s-1)接近,而沿A1分子长短轴进行π扩展得到的两类分子的各向异性均增强.最明显的分子是沿A1分子短轴π扩展的A2分子的各向异性较强;而沿A1分子长轴进行π扩展的A3与A4分子,其各向同性迁移率要优于A2分子,A3与A4分子在Φ=60°和165°方向时具有最大电子迁移率,分别为0.638 和1.59 cm2·V-1·s-1,也具有较好二维传输的电荷传输性质.通过对比A3与A3**的二维各向异性迁移率可知,酰亚胺取代位置不同对该系列分子二维各向异性迁移率的影响.其中,五元环的酰胺取代基团(A3**)的各向同性明显更优,但这是由于其路径1的转移积分相比A3分子极大的降低.综上所述,π扩展会带来各向异性的增加,但长轴π扩展相比于短轴π扩展各向同性要更好.
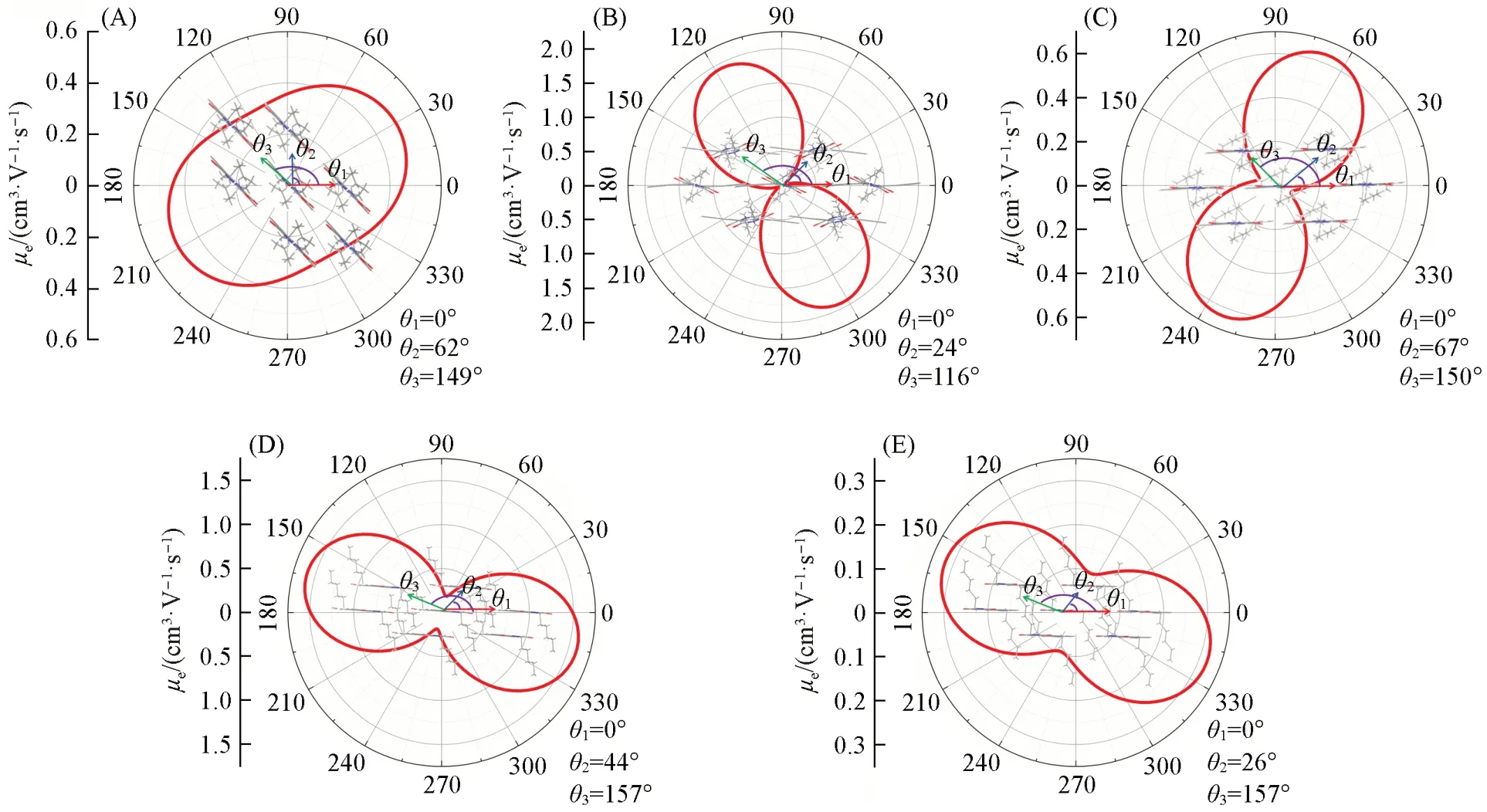
Fig.8 Simulated 2D anisotropic electron mobilities of A1(A),A2(B),A3(C),A4(D) and A3**(E)
3 结论
利用量子化学计算方法研究了基于萘四甲酰基二酰亚胺及其沿分子长短轴π扩展得到的一系列NDI衍生物的分子几何和电子结构、重组能、转移积分、迁移率以及二维各向异性迁移率等性质,并结合分子间相互作用探究了沿萘四酰亚胺不同方向进行π扩展得到的分子结构与性质之间的内在联系.结果表明,短轴π扩展的A2分子的EA值相对更大,具有更好的空气稳定性.此外,引入π扩展可以有效降低分子的电子重组能(约降57~98 meV),但长短轴π扩展降低重组能的途径不同,沿短轴π扩展得到A2有效抑制了高频区内的振动,进而降低重组能;而沿长轴π扩展的A3以及A4不仅降低了高频区的重组能,还有效抑制了低频区内的振动.通过SAPT能量分解,发现A2分子的交换排斥项的占比远高于其它分子,这也导致A2分子堆积形式转变为一维π堆积,并且沿短轴进行π扩展会增加沿短轴的滑移距离,而沿分子长轴π扩展会降低沿短轴的滑移距离,增加沿着长轴的滑移距离.因此,A2分子的路径1的电子转移积分急剧增加而其它传输通道均降低,而长轴π扩展的A3和A4的分子间电子耦合略微降低,其中,A4分子的路径2由于降低了分子间垂直距离而带来了较大的电子耦合.在系列分子中,A4不仅具有较大的二维平均电子迁移率,高达0.15 cm2·V-1·s-1,且其二维各向同性也较好.表明基于萘四甲酰基二酰亚胺,A4的长轴π扩展的方式将最有利于n型电荷传输性质的提高.本文分析的几种不同π核扩展的NDI基n型有机半导体材料的分子结构-分子堆积模式-电子传输性质之间的关系,为设计稳定的高性能电子传输材料提供了一定的设计思路.
