机器学习在高熵电催化材料中的研究进展
2023-12-17宗宇杨李俊辉朱向东单光存马汝广
宗宇杨, 李俊辉, 朱向东, 单光存, 马汝广
(1. 苏州科技大学材料科学与工程学院, 江苏 苏州 215009;2. 北京航空航天大学仪器光电工程学院, 北京 100191)
20 世纪70年代末80年代初, Cantor 等[1]和Yeh 等[2]分别突破了主元概念的传统界限,研发了高熵合金(high-entropy alloy, HEA), 也称为多主元合金(multi-principal component alloy, MPEA). 除了金属体系, 研究人员还开发了高熵陶瓷(high-entropy ceramic, HEC), 包括高熵金属硼化物、高熵碳化物、高熵硫化物、高熵氧化物、高熵氟化物和高熵铝硅化物等.这类含有5 种及以上元素, 并以等摩尔或近摩尔比相互固溶而得到的具有单一相的材料, 统称为高熵材料(high-entropy material, HEM)[3]. 高熵材料的设计策略强调所有参与元素集中在一起, 没有明显的基础元素, 通过各种元素的协同作用形成较好的整体性能[4]. 高熵材料具有独特的4 种效应: 高熵效应、晶格畸变效应、迟滞扩散效应和鸡尾酒效应(见图1). 高熵材料不仅具有成为结构材料的强度和稳定性, 也有作为功能材料的良好性能, 在储能和催化领域具有较大的应用潜力[5].
高熵材料的组成灵活性允许材料性能的微调, 而高熵混合提供了工作条件下的结构稳定性. 例如, 最先引起关注的高熵合金与传统合金相比, 具有较好的机械性能、热力学稳定性、电磁特性以及催化活性等. Qin 等[6]总结了几种高熵合金的合成与应用, 指出高熵合金具有降低电催化剂中贵金属用量的效用. 因此, 高熵材料作为替代贵金属电催化剂的候选材料, 在能源储存与转化方面具有较好的前景. 不可否认, 高熵材料作为电催化剂也存在一些缺点, 如较低的比表面积限制了其活性位点的暴露[7], 且高熵材料作为电催化剂的活性机制尚不清楚, 需要进一步详细研究[8].
高熵材料的多组分特征和多种组合可能性, 使研究人员很少再利用传统方法进行研发. 同时, 高熵电催化材料的活性位点较复杂, 为揭示材料的组分-结构-性能关系也带来了较大挑战[9-10]. 随着计算机硬件和算法的发展, 机器学习(machine learning, ML) 成功地完成了分类、回归、聚类和降维任务, 在大数据归纳、图像和语音识别分类、日常的电子邮件过滤方面都表现出了超强的能力[11]. 机器学习作为处理复杂数据的强大工具也引起了材料科学领域的研究人员的密切关注[12-14]. 基于材料数据库, 机器学习针对特定材料属性建立模型, 快速实现对材料性能的预测, 有望加速新材料设计进程, 缩短材料研发周期[15-17].
本工作主要关注机器学习在高熵电催化材料设计方面的最新进展, 挖掘高熵材料性能与组分、结构等因素之间的关联, 寻找具有物理意义的描述符, 为材料设计提供参考和指导.
1 机器学习
1.1 机器学习的定义
机器学习在Mitchell 等[18]的书中被定义为利用经验改善系统自身的性能, 是一种针对特定的任务目标(target) 来学习经验(experience)、提升性能(performance) 的算法. 机器学习涉及概率论、统计学、近似理论和诸多复杂算法, 依靠对大量数据进行分析并将现有内容进行知识结构划分, 进而高效率地得到预测结果[19].
近年来, 研究人员研发的机器学习方法种类繁多, 考虑强调因素可以有不同的分类方法.目前, 主流的分类偏向强调模型的学习方式, 将机器学习分为如下3 种.
(1) 监督学习(任务驱动型). 输入数据中有标签信号, 以概率函数、代数函数或人工神经网络为基函数模型, 主要采用迭代计算方法在带标注训练集中进行学习, 如决策树(decision trees)、支持向量机(support vector machine, SVM)、集成方法(ensemble method)、朴素贝叶斯分类(Naive Bayes classification)、普通最小二乘回归(ordinary least square regression)、逻辑回归(logistic regression).
(2) 无监督学习(数据驱动型). 输入数据中无标签信号, 主要采用聚类与降维2 种方法,寻找未标注数据中隐含结构, 如聚类(clustering)、主成分分析(principal component analysis,PCA)、奇异值分解(singular value decomposition)、独立成分分析(independent component analysis).
(3) 强化学习(从错误中学习). 以奖/惩信号或环境反馈为输入, 偏重于智能体与环境交互的一种学习方法, 如著名的AlphaGo[20], 在没有任何先验数据的情况下, 通过接收环境反馈来获取学习信息并更新模型参数.
1.2 材料领域中常用的机器学习算法
1.2.1 线性回归算法
线性回归(linear regression, LR) 算法是机器学习中最基础的一类监督学习模型. LR 需要处理的一类问题为给定一组输入样本和每个样本对应的值, 在要求的误差范围内找出目标值和输入值之间的函数关系, 以便于预测任意样本的目标值[21]. 一般形式如下:
式中:{xij|1 ≤i≤n,1 ≤j≤m}是输入样本,{yi|1 ≤i≤n}是样本对应的值. 而系数向量β=(β0,β1,···,βm) 可以通过许多方法求得, 如使用最小二乘法对数据集的均方根误差(root mean square error, RMSE)进行限制而求出.
1.2.2 支持向量回归算法
支持向量回归(support vector regression, SVR) 算法作为SVM 算法的一个分支从而被提出. SVR 本质上是利用核函数将低维数据映射到高维, 然后求解凸二次规划的最优化问题[22]. SVR 与SVM 的区别在于前者主要应用于回归问题, 后者主要应用于分类问题. SVR是一种二分类模型, 先在线性函数f(x) 两侧制造间距为ε(也叫容忍偏差, 为人工设定的经验值) 的间隔带, 然后通过最小化总损失和最大化间隔来得出优化后的模型(见图2(a)).
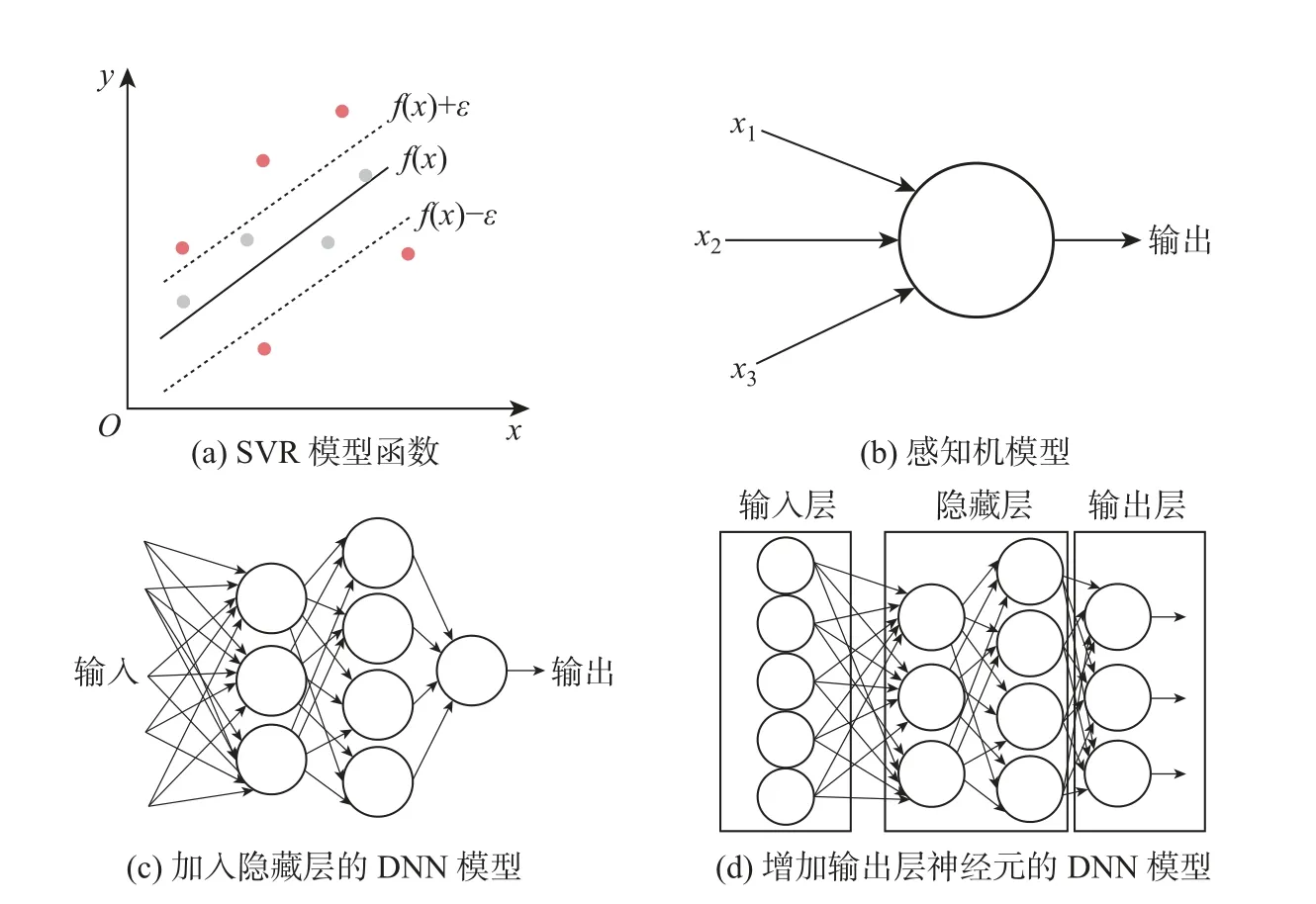
图2 常用的机器学习算法Fig.2 Common machine learning algorithms
SVR 的一般形式如下:
式中:x为样本;w为权重;b为偏置. SVR 对所有落入间隔带内的样本点不计算损失, 即只有支持向量才会对函数模型产生影响. 每个样本点的损失函数如下:
因此SVR 的优化目标函数可表示为
式中: 第1 项为函数f(x)-ε与f(x)+ε的几何距离的倒数; 第2 项为模型的总损失;C为修正系数, 即在间隔带上加入损失, 允许间隔带外存在点, 但这些点带来的损失应尽可能小.
在现实任务中往往很难直接确定合适的ε, 以确保大部分数据都能在间隔带内. 这时可以加入松弛变量δ, 使函数的间隔要求变的宽松, 将部分在间隔带外的点视为在间隔带内.
1.2.3 深度学习算法(以深度神经网络为例)
深度神经网络(deep neural network, DNN) 是深度学习算法中的重要基本模型[23]. DNN是一类多层全连接的神经网络,其大致结构是由一个输入层(input layer)、多个隐藏层(hidden layer) 和一个输出层(output layer) 组成, 其中每一层的各个节点都连接到下一层中的各个其他节点. 根据实际问题的需求可以通过不断增加隐藏层的数量, 使网络变得更深. DNN 也是目前十分先进的一类机器学习模型, 可以模拟许多复杂的问题. DNN 是基于感知机的扩展, 故有时也将其称为多层感知机(multi-layer perception). 感知机是一个输入若干样本、输出一个值的模型(见图2(b)).
输入和输出之间通过构建线性关系, 得到中间输出结果:
接着通过一个神经元激活函数
得到想要的结果1 或者-1.
该模型只能用于二元分类, 且无法学习比较复杂的非线性模型, 因此在工业界无法广泛应用. 而DNN 则在此基础上做了扩展, 可概括为如下3 点.
(1) 加入了隐藏层. 隐藏层可以有多层, 以增强模型的表达能力(见图2(c)).
(2) 增加输出层神经元的数量. 输出层的神经元可以有多个输出, 这样模型便可以灵活地应用于分类回归, 或其他机器学习领域, 如降维、聚类等(见图2(d)).
(3) 对激活函数进行扩展. 感知机的激活函数sign(z) 虽然简单但是处理能力极其有限, 因此DNN 中一般使用其他的激活函数, 如在Logistic 回归中使用Sigmoid 函数:
通过使用不同的激活函数, 进一步增强DNN 的表达能力.
1.2.4 随机森林模型
随机森林(random forest, RF) 是一种比较新的机器学习模型. Breiman[24]提出了分类树算法, 通过反复二分数据进行分类或回归, 大大降低计算量. 2001年Breiman 把分类树组合成RF, 即在变量(列) 和数据(行) 的使用上进行随机化, 生成很多分类树, 再汇总分类树结果.RF 在运算量没有显著增加的前提下提高了预测精度. RF 对多元共线性不敏感, 结果对缺失数据和非平衡数据比较稳健, 可以较好地预测多达几千个解释变量, 被誉为当前最好的算法之一. RF 大致由如下3 个部分组成.
(1) 数据随机选取. RF 通过自扩展(bootstrap) 采样法, 从原始训练样本集N中有放回地重复随机抽取k个样本生成新的训练样本集, 并由此构建k个决策树分类器. 当N充分大时, 训练集中每个样本未被抽中的概率将收敛于36.8%, 即原始数据中约有36.8% 的样本不会出现在Bootstrap 样本中, 这些数据被称为袋外数据, 可用来对模型的误差进行估计.
(2) 分枝方法的选取. 分枝优度准则是基于离均差平方和. 假设有n个自变量X= (x1,x2,···,xn) 和连续性因变量Y. 对于决策树某一节点t的样本量N(t) 可以计算该节点的SS. 假定该节点所有的分枝集合为A,A中的任意元素(分枝) 可以将节点t分为2 个子节点, 记为Lt和Rt. 最佳分枝即是使分枝前后的SS 相差最大的分枝, 即分裂后效果优于分裂前, 使各子节点内的变异最小.
(3) 待选特征随机选取. 与数据集的选取相同, 子决策树的每一分裂过程并未用到所有待选择的特征, 而是从所有待选特征中随机选取一定数量的特征, 然后在选中的特征中挑出最优特征. 这样, 就最大程度地保证每一颗决策树的不同, 提升系统多样性, 从而提高性能.
在寻找最佳分类特征和阈值时目标函数为
式中: Gini 为基尼指数, 用来表示节点纯度, Gini 越大则纯度越低. 如果属性被分为n类, 第i类在当前节点数据集中的占比为p(i), 那么基尼指数的计算方法为
故寻找最佳分类的评判标准可以理解为寻找最佳特征f, 使得当前节点的Gini 值减去左子节点Gini 值和右子节点Gini 值最大.
1.3 机器学习在材料科学中的应用
1.3.1 材料数据库的建立
随着材料基因组概念的不断深化, 全面准确地表征材料组分-结构-性能间的关系是研究和开发新材料的关键, 特别是对于具有庞大排列组合的高熵材料催化剂.
通过数据库可以高精度地建立模型, 预测未知催化剂的催化性能, 了解结构-性能关系, 再使用合适的通用描述符, 可以准确、全面地表示催化剂的结构信息. 同时, 一个有效的描述符可以加速大数据模型的发展, 揭示催化过程的基本物理性质, 掌握材料的本质特征, 更好地应用甚至实现真正的材料按需设计[25-26].
目前, 已有的研究虽然还处于依靠有限的数据进行探索的阶段, 但是也证明了数据驱动型材料科学的研究是非常有效的. 随着全世界对材料学数据库的不断重视、整合和完善, 越来越多的数据资源可供使用, 许多不同类型的材料数据(如物理、化学、机械、电子和热力学) 都可以由包括以量子力学为基础的密度泛函理论(density functional theory, DFT)、Hatree-Fork方法或实验测量(如电导率、离子导率) 生成[27-28]. 如此大的数据为数据驱动技术或机器学习方法的应用提供了机会, 从而加速新的先进材料的发现和设计. 不过, 许多包含大量材料结构和特性的公开数据库, 如Materials Project、The Inorganic Crystal Structure Database(ICSD) 等, 主要是由有序或简单的结构构建的, 对于一个包含化学无序的高熵物质, 目前这些数据库仍然是不可通用. 高熵材料数据库的不足也可以部分归因于其首次发现的时间较短.
1.3.2 材料设计的模型算法
当前, 计算机辅助分子设计(computer aided molecular design, CAMD) 方法被提出并得到了显著发展, 其目的是合理地选择或设计具有指定特性的分子. CAMD 方法自出现以来,已被用于设计溶剂、药品和消费品、工作流体、聚合物、制冷剂和过渡金属催化剂等[29]. 与CAMD 问题类似, 材料的设计任务可以如下定义: 给定一个从实验和(或) 第一性原理计算获得的数据集, 确定具有最佳特性的材料结构和成分.
对于材料设计, 关键的步骤是建立一个相关模型. 该模型可以基于给定的数据集{材料→性质}, 准确描述输入的特定材料的特征(通常为结构特征) 与感兴趣的特性之间的关系. 经典模型的构建在很大程度上依赖于物理观点和机制, 如使用守恒定律和热力学来从已有的参考数据中导出参数(通常为线性或拟合线性) 的数学公式. 机器学习则采用了不同的途径, 即不再依赖原理或物理知识, 而是根据现有的可用数据, 以灵活且非线性的形式训练模型.
图3 展示了基于机器学习的材料发现和设计的通用工作流程[30], 该流程包括3 个主要步骤: 描述符生成和降维、模型构建和验证、材料预测和实验验证. ①用1 组描述符或特征在数据集中表示材料, 需要有关材料和应用程序的特定领域知识; ②在1 组参考材料的已知数据的基础上, 在描述符和目标属性之间建立映射模型, 从简单的线性和非线性回归到高度复杂的核岭回归和神经网络, 各种机器学习方法都可以用来建立这种映射; ③根据所建立的机器学习模型进行反向设计, 以找到具有期望性质的新材料, 合成最佳的候选材料, 并对其真实特性或性能进行实验验证.
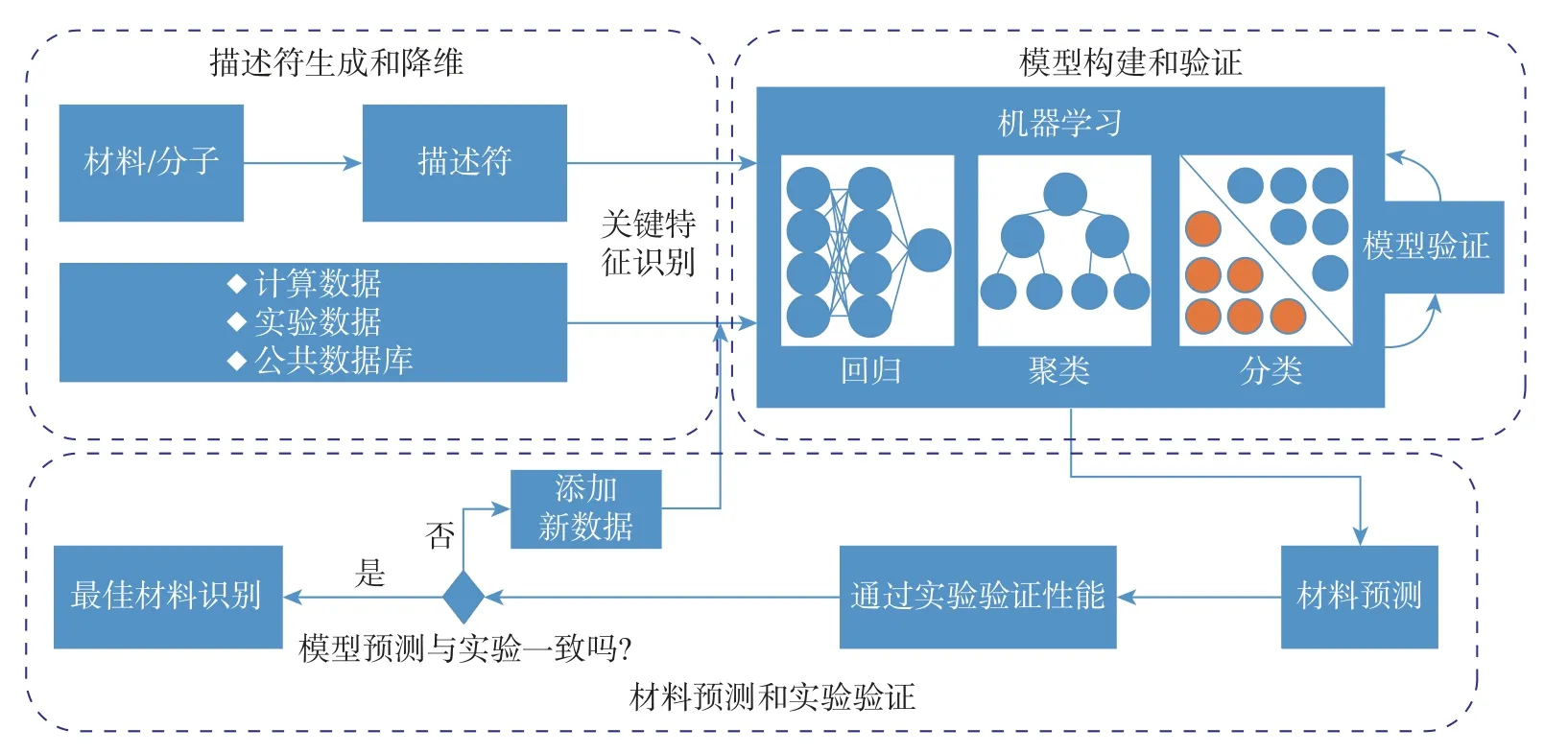
图3 基于机器学习的材料发现和设计的通用工作流程Fig.3 General workflow of materials discovery and design based on machine learning
1.3.2.1 描述符生成和降维
对于材料设计而言, 数据主要来源包括抽样测试的方法和公开的数据库. 抽样测试方法即在构建数据全集后从中以某种原则进行子集抽样并对子集中的数据实施性能测试(理论计算或者实验). 构建数据全集主要提取材料的特征性能, 并把其抽象为描述符.
通常, 机器学习应用程序使用3 种类型的特征描述符: 几何、电子和活性. 当性质来自于体系的几何结构时, 描述符通常被称为结构描述符, 包括原子半径/共价半径、原子序数(即质量数)、基团数、摩尔体积、晶格常数、旋转角度、键长、配位数、活性位点和表面性质(即缺陷/微观结构/界面)[31]. 其出发点是从催化剂的结构特征出发, 构建这些属性与催化性能的关联. 此外, 如果这些性质是由电子密度推导出来的, 则描述符被称为电子描述符[32]. 这些描述符通常从电子结构计算中获得, 即需要花费较长时间通过第一性原理计算, 涉及d 带轨道、带隙、s 带电子、电荷/电荷差、价电子等. 对于过渡金属来说, 主要的反应活性在于d 带轨道, 其性质包括费米能级的中心、填充、宽度、偏度、峰度和密度, 过渡金属在电催化中起着至关重要的作用[33]. 电子描述符较之于结构描述符, 能够更直接地体现不同催化剂与反应中间体之间相互作用程度的差异, 并且其可推广性较之于结构描述符更强. 另外, 还有一种类型的描述符, 用于描述接受或失去电子/质子/基团的能力, 以表明活性, 称为活性描述符, 包括吸附能、电负性、电子亲和度、电离能等[34]. 吸附能反映了基团在电催化剂表面的吸附能力, 可以作为一个描述符来预测电催化的性质(如起始电位、转换频率和产物选择性). 因此, 在运用机器学习过程之前, 识别与所关注的材料特性密切相关的关键特征或描述符始终是至关重要的步骤.
此外, 根据所研究的问题或性质, 可以在不同的复杂度上定义描述符. Sarker 等[35]总结了几个先前已经被开发的重要材料描述符. 最简单的描述符是一维(1D) 参数, 如分子体积、重量和表面积、电子数量和极性. 这些描述符很少包含关于材料或分子实际结构的信息. 在预测某些属性时, 更可取的是使用表示2 维(2D) 甚至3 维(3D) 结构的描述符. 拓扑描述符考虑分子或材料的2 维图形结构, 从而反映对称性、分支和原子连通性等特征. 最常用的拓扑描述符是邻接矩阵和分子连接性指数. 这些描述符的局限性是不包含任何立体化学信息. 一个重要的3D 材质描述符是径向分布函数(radial distribution function, RDF)[36]. RDF 通常由g(r)表示, 定义了在另一个标记的粒子或原子的r距离处找到粒子或原子的概率, 这种类型的描述符可以从实验测量(如X 射线测量) 中获得. 在当前数据库中, 可用的材料数据往往彼此高度相关. 因此, 在构建机器学习模型之前, 有必要使用尺寸缩减工具预处理高维数据集[37]. 有几种算法可为机器学习模型减小特征空间的维度, 并帮助识别最相关的描述符(或关键特征), 如PCA、多维缩放(multi-dimensional scaling, MDS) 和线性判别(linear discriminant analysis,LDA). 例如, PCA 使用正交变换将一组相关变量转换为一组简化的不相关的新变量或主成分(principal component, PC), 在选择每个PC 时应尽量保证与其他PC 不相关, 构成一个可以代表原始数据的缩小的维度空间, 这样信息损失极小.
1.3.2.2 模型构建和验证
机器学习模型的构建在材料发现和设计预测结果中起着关键作用. 一个合适的模型不仅可以保证最终输出结果的可靠性, 也能够大量减少训练需要花费的时间. 总体而言, 在材料设计中分为监督学习、无监督学习2 大类.
监督学习旨在找到一组输入数据映射到相应输出属性的函数, 使用预先标记的数据来学习输出Y和输入X之间的关系, 并且在必须告知Y的值和相应的X值的意义上进行监督.例如,k-最近邻(k-nearest neighbor,k-NN) 算法[38], 其基本原理是通过特征空间中k个近邻的大多数来识别样本, 使用投票机制来处理回归和分类问题.k值的选择以及样本与训练数据在特征空间中的距离是最需要关注的2 个问题: ①k值需要研究人员根据原始训练数据集的特征, 通过交叉验证进行分配; ②k值可以度量模型的复杂度,k值越小表示模型越复杂, 过拟合情况越容易发生. 此外, 如果使用该方法训练的数据集很大, 则k-NN 的预测非常耗时, 并且内存占用也很大, 训练数据的不平衡也会影响k-NN 的性能.
无监督学习则在没有任何事先指导的情况下学习数据的属性, 如通过根据数据的特征将数据分组或通过在高维空间中找到数据变化的主导方向[39]. 由于每种方法或算法都有其自身的适用性和适用范围, 因此选择合适的机器学习算法对于其成功实施至关重要. 最小二乘回归、核岭回归、神经网络和决策树这几种算法都可以创建属性预测模型. 但是, 某些算法(如基于回归的算法) 提供了实际的预测功能, 而其他算法(如决策树) 则没有. 此外, 可用数据的数量也决定了学习算法的选择. 例如, 要正确处理数十至数千个数据点, 可以使用诸如克里金和核岭回归的回归方法, 但是当数据比这大得多时, 则需应用更复杂的学习方法, 如深度神经网络.
对于机器学习模型而言, 不仅要求其对训练数据集有很好的拟合(训练误差), 同时也希望对未知数据集(测试集)有很好的拟合结果(泛化能力),所产生的测试误差被称为泛化误差[40].度量泛化能力的好坏, 最直观的表现就是模型的过拟合(overfitting) 和欠拟合(underfitting).过拟合和欠拟合是用于描述模型在训练过程中的2 种状态. 一般来说, 训练过程会是如图4 所示的一个曲线图.

图4 欠拟合与过拟合示意图Fig.4 Schematic diagram of underfitting and overfitting
刚开始训练的时候, 模型还在学习过程中, 处于欠拟合区域(指模型不能在训练集上获得足够低的误差), 此时模型复杂度低, 模型在训练集上表现较差, 没学习到数据背后的规律. 随着训练的推进, 训练误差和测试误差都下降. 在到达一个临界点之后, 训练集的误差下降, 测试集的误差上升了, 这时就进入了过拟合区域(指训练误差和测试误差之间的差距太大), 模型复杂度高于实际问题, 模型在训练集上表现较好, 但在测试集上却表现较差, 泛化能力差, 从而对训练集以外的数据预测不精确.
综上, 欠拟合基本上都会发生在训练刚开始的时候, 经过不断训练后欠拟合能得到解决.如果此时还存在欠拟合, 则可以通过增加网络复杂度或者在模型中增加特征, 来解决欠拟合的问题. 要想解决过拟合问题, 就要显著减少测试误差而不过度增加训练误差, 从而提高模型的泛化能力. 可以使用正则化(regularization) 方法[41]修改学习算法, 使其降低泛化误差而非训练误差.
常用的正则化方法根据具体使用策略的不同可分为①直接提供正则化约束的参数的方法, 如L1/L2 正则化; ②通过工程上的技巧来实现更低泛化误差的方法, 如提前终止(early stopping) 和暂退法(dropout); ③不直接提供约束的隐式正则化方法, 如数据增强等.
(1) 获取和使用更多的数据(数据集增强)——解决过拟合的根本性方法.
使机器学习或深度学习模型泛化能力更好的办法就是使用更多的数据进行训练. 但是,在实践中拥有的数据量是有限的. 解决这个问题的一种方法就是创建假数据并添加到训练集中——数据集增强. 通过增加训练集的额外副本来增加训练集的大小, 进而改进模型的泛化能力.
(2) 采用合适的模型(控制模型的复杂度).
过于复杂的模型会带来过拟合问题. 对于模型的设计, 目前公认的一个深度学习规律是越深入越好. 对于卷积神经网络(convolutional Neural Networks, CNN) 来说, 层数越多效果越好, 但是也更容易产生过拟合, 并且计算所耗费的时间也越长. 而根据奥卡姆剃刀法则, 应该选择简单、合适的模型解决复杂的问题.
(3) 降低特征的数量.
对于一些特征工程而言, 可以降低特征的数量, 删除冗余特征, 人工选择保留特征来解决过拟合问题.
(4) 暂退法.
暂退法是在训练网络时用的一种技巧(trick), 相当于在隐藏单元增加了噪声. 暂退法指的是在训练过程中每次按一定的概率(比如50%) 随机地删除一部分隐藏单元(神经元). 暂退法目的是在训练过程中产生不同的训练模型, 这些不同的训练模型也会产生不同的计算结果. 随着训练的不断推进, 计算结果会在一个范围内波动, 但是均值却不会有很大变化, 因此可以把最终的训练结果看作是不同模型的平均输出, 并且消除或者减弱了神经元节点间的联合, 降低了网络对单个神经元的依赖, 从而增强了泛化能力.
(5) 提前终止.
对模型进行训练的过程即是对模型的参数进行学习更新的过程, 这个参数学习的过程往往会用到一些迭代方法, 如梯度下降(gradient descent). 提前终止是通过迭代次数截断来防止过拟合的方法, 即在模型对训练数据集迭代收敛之前停止迭代来防止过拟合.
为了获得性能良好的神经网络, 训练过程中可能会经过很多次轮数(epoch). 而提前终止就是在每个轮数(或每N个轮数) 结束后, 在验证集上获取测试结果. 随着轮数的增加, 如果在验证集上发现测试误差上升, 则停止训练, 将停止之后的权重作为网络的最终参数.
1.3.2.3 材料预测和实验验证
如图3 所示, 在建立机器学习模型后, 可以通过交叉验证法来评估模型的稳健性和泛化性[42]. 将大小为k的训练样本划分成大小为k-1 的校准样本和大小为1 的验证样本, 重复k次. 划分验证集, 对训练集生成的参数进行测试, 从而相对客观地判断这些参数与训练集外数据的符合性, 选择最优模型. 交叉验证方法评价结果的稳定性和保真度在很大程度上取决于k的值, 因此交叉验证方法通常被称为k-fold 交叉验证, 其中k最常用的值为5 和10. 留一交叉验证(leave-one-out cross validation, LOOCV) 方法是在数据集较少的情况下交叉验证的一种特殊形式, 即只使用原始训练集中的一个样本作为验证集, 其余样本作为训练数据. 对于小数据集的情况, 另一种有用的方法是自举法(bootstrap), 该方法可以通过带替换的抽样方法从初始数据集生成所需大小的训练集. 然而, 由自举方法生成的数据集的分布与初始数据集的分布不同, 这将引入估计偏差. 因此, 当数据量足够时, 通常采用交叉验证方法. 在对模型进行评价时, 除了对模型的预测性能进行评价外, 还应考虑模型的效率、复杂性、稳健性和可移植性.
当机器学习模型通过验证后可以进行反向设计以根据模型查找具有所需特性的材料. 该过程通过使用大规模筛选或数学优化来完成. 大规模筛选方法的基本思想是, 在设计空间中生成所有可能的候选材料后使用已学习的模型逐一进行测试. 通常, 材料的生成必须考虑对材料的几个限制, 这些限制通常以2 种形式存在: 结构和组成成分. 因此, 需要使用一个系统的程序来识别设计空间中的所有材料(或尽可能多的材料). 在生成候选材料后, 使用经过训练的模型可简单、直接地评估其属性, 或者可以将反向材料设计公式转化为数学优化问题, 其中目标特性在受到结构和成分约束的情况下得到优化. 优化的方法试图在不测试设计空间中所有候选对象的情况下确定有前途的材料, 这使该方法受组合复杂度的限制要小得多. 确定性或随机算法均可用于解决所确定的最优材料的成分优化问题. 在确定最佳材料后, 就能够合成这些材料并通过实验验证其实际性能. 如果实验结果与预测结果吻合良好, 则可以证明该模型对于该问题拟合预测效果良好, 通过该模型可以继续探索新催化剂; 如果实验结果不符合预测, 可将所设计的材料及相应的实验结果添加到训练集中, 并重新训练机器学习模型.
2 高熵材料
2.1 定义
含有5 种或5 种以上的等摩尔比合金形成的单一固溶体被定义为高熵合金(high-entropy alloy, HEA)[1,37], 也有人认为不应该限制元素浓度[43-44]. 此外, 合金的熵可由构型熵公式来计算:
式中: ∆Smix为混合熵;R为摩尔气体常数;ci为第i元素的摩尔分数. 因此, 根据合金的熵值大小, 可将合金分为低熵合金(low-entropy alloy (LEA), ∆Smix< 1.0R)、中熵合金(medium-entropy alloy (MEA), 1.0R< ∆Smix< 1.5R) 和高熵合金(HEA, ∆Smix> 1.5R).这种材料的结构无序程度将明显大于二元或三元合金, 从而产生更高的熵, 有更高的热力学稳定性. 由式(10) 得到构型熵后可以得到一系列摩尔比相等的高熵合金(见图5(a))[45].
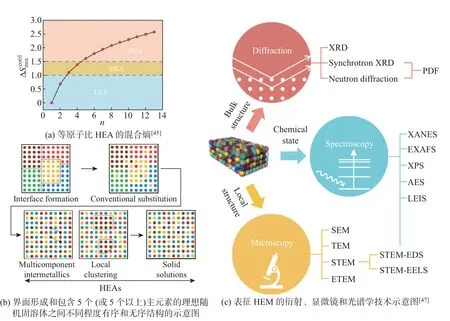
图5 高熵材料的定义、结构特点和表征方法Fig.5 Definition, structure feature and characterization techniques of high-entropy matorials
高熵化合物, 也称为高熵陶瓷, 通常包含了除金属元素外的其他非金属元素(如氢、氧、硫、氟等), 与金属化合物框架的前驱体一起, 通过沉淀、加热、机械加工等各种方法形成稳定的高熵化合物, 具体定义相似于高熵合金[46].
2.2 特点
高熵合金的晶体结构通常为简单的面心立方(face-centered cubic, FCC) 结构、体心立方(body-centered cubic, BCC) 结构以及密排六方(hexagonal close packed, HCP) 结构, 不同原子随机占据晶格位置, 形成简单固溶体. 目前, 已报道的高熵合金纳米催化剂最常见的结构为FCC, 其他结构如BCC 和HCP 结构, 局部序度的改变会造成键长的变化从而带来结构上的特殊性(见图5(b)). 另外, 包括活性位点的定性和定量、机理研究和催化性能的优化在内的研究都依赖于高熵材料的精确表征. 由于高熵材料成分和结构的复杂性增加, 故这是具有挑战性的. 当然, 尽管存在挑战, 利用各种技术来表征高熵材料已经取得了很多进展(见图5(c)).
基于高熵合金的结构, 研究人员总结出高熵合金不同于传统合金的特征, 即所谓的4 个核心效应, 即高熵效应、晶格畸变效应、迟滞扩散效应和鸡尾酒效应[47].
高熵效应由高熵合金早期的概念而来, 即5 种或5 种以上元素均匀混合, 合金体系能够获得较大的混合熵, 使合金体系更倾向于形成固溶体, 而不是金属间化合物, 该特性体现了混合熵对合金相形成的贡献[48]. 根据经典Gibbs 相律, 对于一个给定的合金, 当体系压力恒定时,其满足
式中:C为合金中所含元素的个数;P为所形成的相的数目;F为体系的自由度. 已有的研究结果显示, 高熵合金生成相的数目要远小于经典吉布斯相律所预测的合金体系所生成的最大平衡相的数目. 从自由能表达式(G=H-TS) 可以看出, 当合金的混合熵高到可以足够抵消混合焓的作用时将促进固溶体的形成, 特别是在足够高的温度下, 高的混合熵能够稳定均匀混合的固溶体[49]. 因此, 高熵合金中存在的相数明显减小.
晶格畸变效应是由于组成高熵合金的每个组分的原子大小的巨大差异而产生的. 金属原子在晶格中的随机占据会导致严重的晶格畸变. 晶格畸变效应造成同一层原子面的高低不平,使得X 射线在不平整的晶面上产生明显的布拉格散射, 从而导致高熵合金X-射线衍射峰强度弱化及展宽现象[50]. 这种严重的晶格畸变不仅显著提高了高熵合金的硬度, 而且还降低了导电性和导热性, 且原子在表面的扩散受到阻碍, 这种效应也有助于形成纳米级高熵合金.
高熵合金的晶格畸变导致原子在晶格内的扩散激活能增大, 从而降低了原子的有效扩散速率, 形成迟滞扩散效应. Zhang等[51]运用高温扩散偶的方法研究了Mn、Cr、Fe、Co 和Ni 5种原子在近乎理想固溶体结构的CoCrFeMnNi 合金中的扩散行为. 结果表明, 相比于包含相同组元的传统FCC 结构的合金, 各个组元在高熵合金基体中的扩散系数均远小于在其他单组元中的扩散系数. 相应地, 该元素在高熵合金中的激活能也高于参考金属中的活化能.
高熵合金的鸡尾酒效应是指因各元素之间的相互作用而带来的一种协同效应, 即元素的特别组合可能带来合金性质上的特异性[49]. 鸡尾酒效应最初是为了解释金属玻璃、超弹性合金和超塑性合金的特殊性质而引入的. 仅从高熵效应来预测是否形成单一固溶体或判断高熵合金的热稳定性是不够准确的. 另外, 原子的扩散速率与材料的晶粒尺寸有极大关系, 当合金由纳米晶组成时原子迅速转移, 只有在大晶粒条件下原子的扩散速率与所述的迟滞扩散效应才相符. 鸡尾酒效应笼统地解释了各组元之间的协同效应, 但其具体的物理意义尚不明确.
与高熵合金相似, 高熵化合物(高熵陶瓷) 也是通过将多种元素插入晶体中来建立一种热力学稳定. 由于元素种类多样, 当重新排列元素时会导致这些元素产生更规律的秩序, 从而违背了熵增原则. 因此, 高熵化合物往往具备耐高温、抗腐蚀的特性, 同时由于多种元素的协同作用, 故其表面往往具备大量活性位点.
2.3 优势与劣势
与传统催化材料相比, 高熵材料的主要优势是由鸡尾酒效应引起的. 受益于多个合并元素, 高熵材料可以被视为原子复合材料, 通常表现出全新的特性, 并且可以通过更改不同元素的比例来改变特性. 除此之外, 高熵材料功能单元之间非常强的协同效应在贵金属和非贵金属活性中起决定性作用. 相较于传统合金或化合物, 高熵材料因为掺入的金属原子的大小明显不一, 会产生严重的晶格畸变, 这有利于暴露出更多的催化位点, 从而在催化领域展现出潜力.
尽管与传统合金和其他化合物相比, 高熵材料具有固有优势, 但在催化领域的应用中仍然面临一些挑战. 首先, 即使结果表明高熵材料的特性可以通过变更不同元素或比例来调配, 但由于鸡尾酒效应多元素的混合, 单个元素在催化中起的作用仍然较为模糊. 此外, 在工业化应用中寻找一种简单通用的合成高熵材料的方法也是有必要的[12].
总的来说, 到目前为止进行的研究仍处于早期阶段, 大多数的结构裁剪仍然是随机完成的, 因此合理地设计高熵材料非常具有挑战性. 除了广泛开发高熵材料和高熵陶瓷外, 探索新类别的高熵材料也应受到高度关注. 另外, 研究人员需要对高熵概念本身有更深入的理解, 通过类比研究, 删除一种或多种元素来探讨高熵材料和相应的中熵或低熵材料的特点与优势. 这样的实验将提供更多见解, 特别是性能和反应之间的比较机制.
3 机器学习在设计高熵催化剂中的应用
通常, 催化剂的最佳设计都是凭经验或实验实现的. 量子化学计算为使用第一性原理设计催化剂提供了可能性. 然而, 较高的计算成本将其应用限制在相对简单的反应和少量的候选催化剂上. 随着可用的实验和计算数据的迅速增加以及催化信息学的发展, 如今已可以使用机器学习模型很好地描述催化剂的结构和活性之间的关系, 这对于催化剂的开发非常有帮助. 迄今为止, 许多机器学习模型已成功用于预测高熵陶瓷的相形成和力学性能, 如ANN、SVM、RF模型等[52]. 图6(a) 展示了机器学习预测多组分固溶体形成的方法, 主数据集包含由结构建模技术生成并报道的多组分二硼化物, 通过机器学习不同的模型、超参数调优和交叉验证, 获得最终模型, 并预测新的四元、五元化合物[53]. 最近, Zhang 等[54]利用SVM 和ANN 模型预测了单相的碳化物高熵陶瓷, 流程如图6(b) 所示. ANN 模型被广泛应用于深度学习, 而SVM 在处理小的数据集方面具有很大优势; 二者结合能够快速搜寻整个组分空间, 而不再需要昂贵的DFT 计算.当前高熵合金催化剂也已被证明在氨分解反应(ammonia decomposition reaction, ADR)、析氧反应(oxygen evolution reaction, OER)、氧还原反应(oxygen reduction reaction, ORR)、CO2还原反应(CO2reduction reaction, CO2RR) 等方面具有比其他金属或合金体系更好的催化性能[55-58].
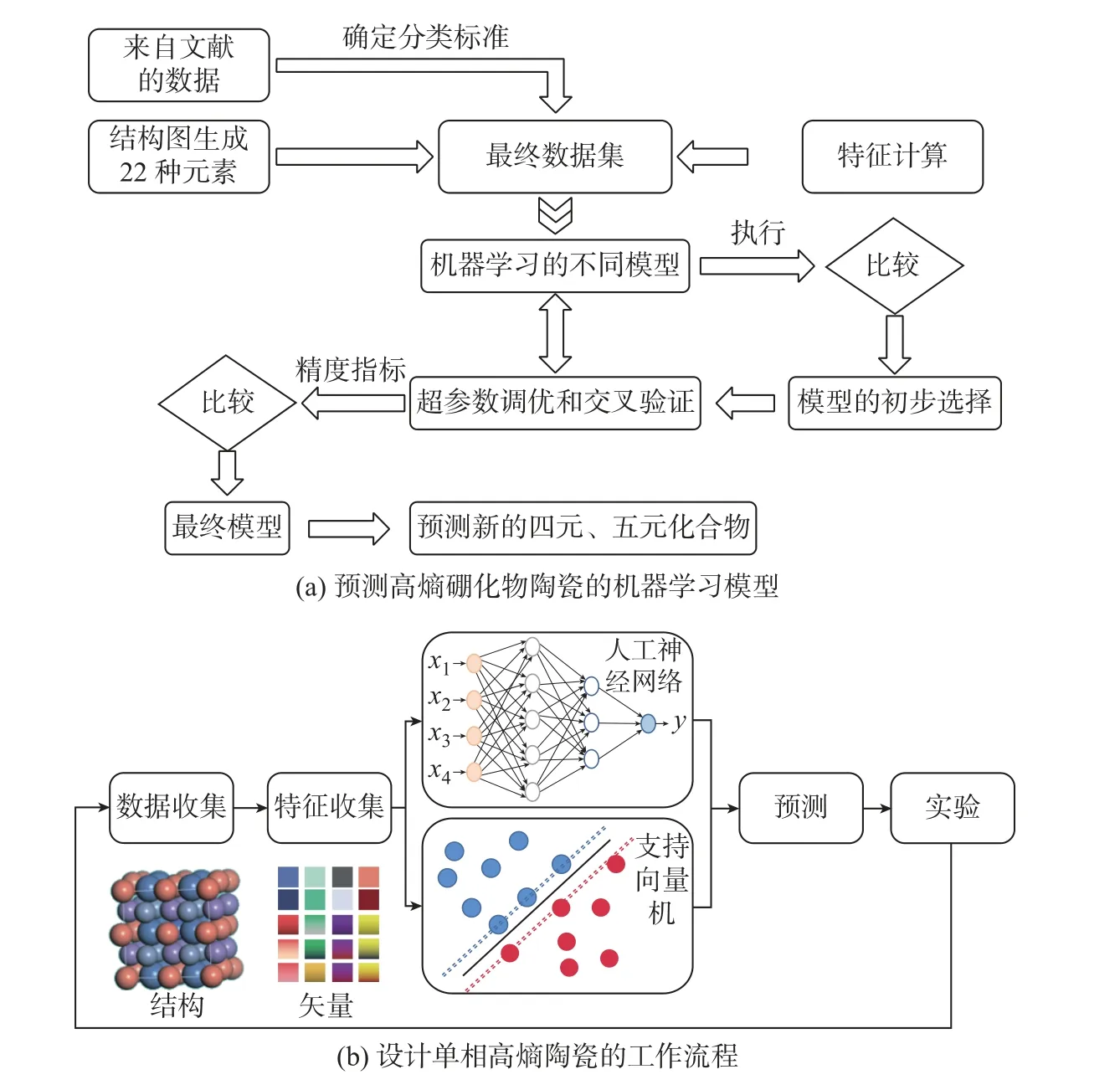
图6 机器学习预测高熵化合物流程Fig.6 Workflow of predicting high-entropy ceramics via machine learning
高熵氧化物(high-entropy oxide, HEO) 是一种新型材料, 在能源和催化领域具有广阔的应用前景. 然而, 一系列的高熵氧化物太新颖, 无法评价其合成性质, 包括形成性质和基本性质. 基于DFT 的第一性原理计算[59-60]广泛应用于物理、化学和材料科学, 是一种量子力学建模方法. 但当计算非化学计量数的化合物时, 其计算时间很长, 计算能力的负荷很大. DFT通过与机器学习结合可以减少计算时间和计算负荷, 其中机器学习利用计算机算法通过DFT经验自动预测物理、化学和材料特性. 例如, Lin 等[61]将Cr、Co、Fe、Mn 和Ni 分配到晶格位置作为自变量, 然后采用DFT 对尖晶石结构的多元素化合物进行了计算, 将得到的尖晶石晶格常数以及形成能作为因变量, 将通过DFT 计算出来的数据作为训练集, 分别使用反向传播网络(back propagation network, BPN) 和遗传算法神经网络(genetic algorithm neural network, GANN) 这2 种算法进行了模拟计算, 并通过使用RMSE 值和散点图来选择和确定合适的模型. 机器学习预测与DFT 计算一致(晶格常数和形成能分别在2% 和1% 的偏差范围内), 这表明该研究中使用的流程和设计是可行的. 此外, 已建立的计算尖晶石结构的晶格常数和形成能的数据库, 可用于了解尖晶石结构高熵材料和其他尖晶石结构氧化物的性质, 为数据密集型材料科学和计算具有Co、Cr、Fe、Mn 和Ni 金属的尖晶石结构材料的特性提供了机会.
3.1 机器学习在HER 催化剂中的应用
氢气作为一种高能量密度的无污染能源而受到广泛关注. 电解水析氢反应(hydrogen evolution reaction, HER) 对氢能转换和储存具有重要意义[62]. HER 是一个典型的双电子转移反应, 中间只有一个H*, 其中* 表示吸附. 双电子转移反应可能通过Volmer-Tafel 机制或Volmer-Heyrovsky 机制发生(见表1). 表中机理是机器学习辅助电催化剂设计的基础, 并已被许多研究人员引用[53].

表1 HER 反应机理Table 1 HER mechanism
多元合金催化剂是提高催化性能的最有前途的方法之一, 其协同性能远超低组元合金预期[63-64]. 然而, 由于金属元素在催化反应中的物理行为和化学活性不同, 确定最佳元素成分和组成具有挑战性. 此外, 由于很难精准地确定金属元素在合金内的催化性能, 故很难确定需研究的金属组合, 以前都是通过模拟计算二三元合金的穷举法[65-66]. Kim 等[67], 通过实验和主动学习相结合的方法, 面向HER 展示了一种可以搜索多金属合金催化剂的最佳成分的方法, 其中该模型主动学习的输入数据(即金属前驱体的成分和组成以及测量的过电位) 来自于多金属合金纳米颗粒的合成及其过电位的实验测量. 该团队训练了一个高斯过程(Gaussian process, GP) 模型来学习一个函数, 该函数将一个由8 维前驱体组成的向量映射到一个标量过电位值. 随着实验模型不断迭代, 只需将前驱体混合物组成作为输入数据, 在不考虑合金的实际成分、催化剂的表面积和负载量、催化剂的阻抗等其他特性作为输入数据的情况下, 主动学习和实验的结果在迭代过程中具有相同的趋势, 能有效预测系统的过电位(见图7). 然后,该团队采用碳热冲击(carbon thermal shock, CTS) 法[68], 在前驱体混合物中加入不同摩尔分数的金属, 合成多金属合金催化剂, 直接预测催化过电位的实验可调参数(见图7(i)∼(ii)). 在完成催化剂的制备后, 通过线性扫描伏安(linear sweep voltammetry, LSV) 法测量电流密度为20 mA·cm-2时的过电位, 表征合成纳米颗粒的催化性能(见图7(iii)). 主动学习以高效的方式找到能够表现出最佳催化性能的前驱体的最佳组成, 使得实验过电位小于纯Pt 催化剂的过电位. 可见, 该方法论具有应用于其他高熵合金催化系统的潜力.

图7 寻找低过电位多金属合金催化剂的整体工作流程Fig.7 Overall workflow for searching multi-component alloys with low overpotential
3.2 机器学习在OER 催化剂中的应用
OER 是电解水的另一个半反应, 同时也是空气电池的负极[69]. 由于四电子反应动力学更为缓慢, 因此OER 往往是总反应中的瓶颈. 表2 对酸性和碱性环境中OER 的机理进行了分析, 表明电催化剂设计的主要挑战之一在于中间体*OH、*O 和*OOH 的吸附能之间存在很强的相关性[70]. 打破相关关系以获得优异的性能是OER 电催化剂设计的主要目标.

表2 OER 反应机理Table 2 OER mechanism
人工智能(artificial intelligence, AI) 和机器学习是达成这个目标的方案选择之一, 是利用统计模型和优化算法揭示训练数据背后的隐藏特征从而进行预测. Rohr 等[71]使用基于四元金属氧化物电催化剂的序列学习(sequence learning, SL) 加速了材料发现过程, 从而量化优异的电催化剂性能和准确性. SL 技术旨在使机器学习模型和获取函数易于变化, 并在表示所有可能实验的离散搜索空间的假设下实现, 将其称为大小为n的样本集. 该集合中的每个样本都由其实验坐标来表示, 故该坐标是6 维组成向量. 该技术可以保证每个实验的优点值, 且这种SL 技术可以使用任何机器学习模型来实现. 该研究团队选择了3 个涵盖了广泛的SL 算法的模型. 线性集成(linear integration, LE) 方法, 作为代表性贝叶斯方法的GP 和RF 模型,选择OER 过电位作为性能指标. 不同的SL 方案在4 种化学成分上进行了测试, 每种化学成分含有2 121 种催化剂(见图8). 相对先进的GP 和RF 算法比LE 算法更出色, 通常GP 和RF 算法大约进行100 次循环就能找到80% 的顶级催化剂, 而LE 算法则需要200 多次循环.在特定情况下与随机获取方法(random choice method, RCM) 相比, 通过SL 算法训练出的模型预测电催化剂的设计速度可以加快20 倍. 此外, 研究还表明, 对催化剂发现的SL 算法进行基准测试可以加速研究, 但还没有达到预期AI 引导发现的数量级. 通过探索3 个互补的研究目标(即发现任何好的催化剂、发现所有好的催化剂和发现一个预测模型) 和3 个互补的机器学习模型(GP、RF、LE), 可以证明SL 算法的性能更多地取决于研究目标而不是模型的种类. 不同组成空间的4 种催化剂数据集模型性能的可变性揭示了一致的定性趋势, 表明观察结果具有一定程度的普遍性.

图8 序列学习预测金属氧化物电催化剂Fig.8 Prediction of metal oxides as electrocatalysts by sequence learning
3.3 机器学习在ORR 催化剂中的应用
在燃料电池等设备的能量转换过程中, ORR 在电催化过程中起着举足轻重的作用, 其阴极缓慢的动力学限制了燃料电池的整体性能[72]. 因此, 加速电催化剂的设计, 以促进燃料电池的ORR 动力学是非常重要的[73]. 目前, 昂贵高效的Pt 基材料是最实用的ORR 电催化剂. 然而, 电催化剂的高成本是燃料电池商业化的一个挑战. 因此, 应用机器学习寻找高效廉价ORR 催化剂至关重要. 在酸性和碱性环境中, ORR 的机制已经得到了较好的研究[73](见表3). ORR 催化剂设计面临的最大挑战在于ORR 反应中间体*OH、*O 和*OOH 的结合能之间存在不利的关系.

表3 ORR 反应机理Table 3 ORR mechanism
机器学习能高效地发现有效的高熵合金ORR 催化剂. 高熵合金中的各种元素以随机分布的组分形成有序的晶体结构, 能提供具有优秀的催化性能的原子排列位点. 多种元素随机分布的过渡金属晶体结构, 使贵金属的负荷降低, 同时提高其性能. Wan 等[9]首先选择了6 种过渡金属元素(Ir、Pt、Ru、Rh、Ag 和Fe), 通过这些元素构建五元的高熵合金来研究其稳定性.由于这些元素具有相似的原子半径和接近的晶格常数, 具有相同的FCC 结构, 故常常被报道用于ORR 催化剂[75-79]. 高熵合金的完全随机构型空间导致的高度无序性, 增加了高熵合金的混合构型熵, 有利于形成稳定的单相固溶体结构而不是易碎的金属间化合物. 根据休谟-罗瑟里规则, 原子半径之差和生成焓与熵之比可以直接评价其稳定性[80-81]. 然后, 根据Sabatier规则[82], 反应中间体的吸附能通常是催化活性的良好描述符, 选取∆GOH∗作为活性描述符建立高熵合金表面反应位点ORR 催化活性与OH* 吸附能之间的ORR 火山曲线. 随机选取具有不同Miller 指数表面和组成元素的高熵合金上的360 个反应位点, 通过DFT 计算得到位点上OH* 中间体的吸附能, 作为机器学习模型的原始数据集. 随后, 通过特征工程的方法将描述位点局部环境的问题抽象为组成位点的2 个金属原子的配位原子特征. 在完成特征工程和数据提取后, 使用预处理后的数据集用7 种不同的回归算法训练7 种不同的机器学习模型:包括梯度增强回归(gradient boosting regression, GBR)、前馈神经网络(feedforward neural network, FNN)、随机森林回归(random forest regression, RFR)、支持向量回归、K近邻回归(Kneighbor regression, KNR)、核脊回归(nuclear ridge regression, KRR) 和最小绝对收缩和选择算子回归(least absolute shrinkage and selection operator, LASSO). 在4 次交叉验证后, 比较测试集上的模型指标, 选择GBR、RFR 和FNN 算法来建立更好的机器学习模型;通过手动调参比较选择了RMSE 较低、决定系数(R2) 评分较高的GBR 模型. 随机等量生成具有不同Miller 指数晶面和组成元素的高熵合金上12 000 个不同的桥点, 即每种高熵合金有2 000 个点, 其中(100) 晶面分为1 000 个点, (111) 晶面分为1 000 个点. 将这些位点输入到性能良好的GBR 模型中, 对输出的吸附能进行即时收集和分类. 对结果的深入分析表明: 高熵合金表面的吸附能是所有环境配位成分金属原子单独贡献的混合; 直接与OH* 键合的2 个金属原子是决定ORR 中间体吸附能的主要因素, 配位原子离OH* 越近其影响越大; 高精度地预测了高熵合金不同晶面上数百万个反应位点的OH* 吸附能; Wan 等[9]对预测结果和机器学习模型进一步分析, 提出了一种提高高熵合金催化剂ORR 活性的策略, 即通过优化表面来扩大找到具有所需OH* 吸附能的高效活性位点的可能性. 整个机器学习过程如图9 所示, 可以看出该设计为高熵合金催化剂的合理设计和纳米结构的合成提供了指导.
与之相同的基于机器学习的方法, 可以通过使用相对较大但可管理的DFT 数据库开发计算效率高的模型来评估催化效率, 并且缓解大量不同的化学环境对确定反应机理带来的挑战.Saidi[83]重点研究了新近发现的PdAuAgTi 合金, 该合金在Ti 成分空间的狭窄区域内表现出较好的ORR 活性[84]. Saidi 假设PdAuAgTi 为具有FCC 晶格的单一固溶体相, 通过计算创建一个大型数据集∆E, 该数据集由合金的随机结构配置生成, 其组分、原子排列和OH 吸附位点各不相同; 利用开发的数据库[85], 使用基于吸附位点几何和化学环境特征的DNN 来训练一个特定位点的机器学习模型. 结果证明PdAuAgTi 合金在8%∼12% Ti 的狭窄区域内与OH 结合最佳, 与鉴定出的11%∼13% Ti 的实验结果一致. 另外, Saidi 还根据合金的*OH 结合强度绘制了整个合金的成分空间. 该研究扩展到PdCuZnTi 合金, 并揭示了小于8% Ti 的成分值范围, 也有较好的ORR 活性. 这种合金将比PdAuAgTi 更具成本效益, 具有取代Pt的潜力. 高通量筛选结合DFT 计算和机器学习的数据来分析Pd 基四元合金氧还原活性的增强机制, 较好地解释了实验结果, 并提供了催化活性在总的组分空间的精确图谱. Saidi 的研究强调了高通量计算和机器学习之间的结合对原子水平上的材料设计的重要影响.
总之, 基于机器学习设计ORR 的电催化剂通常有2 种策略: 寻找替代电催化剂(非贵金属电催化剂) 或降低贵金属的负载量, 其中*OH、*O 和*OOH 的吸附能是电催化剂设计的重要活性描述符.
3.4 机器学习在CO2RR 催化剂中的应用
电催化CO2RR 能产生高附加值的化学品和燃料, 已引起广泛关注, 因为CO2RR 提供了一种清洁有效的方法来缓解能源短缺, 同时减少全球碳排放[86]. CO2的电化学还原方法多种多样, 可产生16 种不同的产物, 包括C1 产物(即CO、HCOOH(甲酸)、HCHO(甲醛)、CH3OH(甲醇)、CH4(甲烷)) 和多碳产物(即H2C2O4(草酸)、CH3CH2OH(乙醇)、CH2= CH2(乙烯)、CH3CH3(乙烷) 和CH3CH2CH2OH(正丙醇)), 各种电子还原反应如表4 所示(注: SHE (standard hydrogen electrode, 标准氢电极)).

表4 CO2RR 的反应、电势(E0 vs. SHE 和pH = 7) 和电子转移数(n)Table 4 Reaction steps, potential (E0 vs. SHE and pH=7) and electron-transferred number (n) of CO2RR
CO2还原反应中间体吸附能之间的标度关系为只考虑一种中间体筛选高活性催化剂提供了一种简单的方法. 然而, 标度关系也限制了CO2RR 催化性能的突破[54]. 因为没有办法同时保证催化剂表面对一种反应物的吸附是强的, 有利于活化; 对其他反应物的吸附是弱的, 有助于产物的形成. 例如, Feaster 等[87]比较了CO2RR 中间体的吸附能趋势, 并基于这一分析提出了一个活性火山. CO* 的强吸附能导致CO* 难以质子化(CO* + H++ e-→CHO*), 而CO* 的弱吸附能使得CO2的质子化过程缓慢(CO2+ H++ e-→COOH*). 当CO* 达到合适的吸附能时, CO2RR 的催化性能达到了火山口(最佳), 但无法进一步提高. 因此, 超越火山口的催化性能并打破这种关系成为研究人员关注的热点.
高熵合金中多元素的协同作用产生了不同范围的吸附位点, 在不施加外力或引入复杂界面结构的情况下提供了规避催化中耦合关系的机会. Chen 等[56]采用DFT 方法探索了Fe-CoNiCuMo 的高熵合金体系用于电催化CO2RR. 该团队首先采用Fe0.2Co0.2Ni0.2Cu0.2Mo0.2最稳定的表面(111) 作为CO2RR 的活性表面; 然后, 使用神经进化结构(neuro evolutionary structure, NES) 方法生成了200 个FeCoNiCuMo (111) 结构, 并在这些生成的结构中, 选择了20 个结构来计算CO2RR 过程中一些重要中间体的吸附. 由于CO2和CO 的高稳定性, 故在CO2RR 过程中CO2或CO 的活化通常是潜在的电势限制步骤(potential limiting step,PLS). 因此, 该团队考虑了CO2(CO2+H++e-→COOH*) 和CO(CO*+H++e-→CHO*)的质子化反应, 计算1 280 个吸附位点对COOH*、CO* 和CHO* 的吸附能. 为了确定高熵合金活性中心的标度关系如何消失, 同时计算了活性中心的电子结构, 并在此基础上建立了高精度的神经网络(neural network, NN) 模型来预测相应的吸附能(见图10(a)∼(e)). 在CO2RR过程中存在2 个旋转区域, 分别是COOH* 和CHO* 旋转, 克服0.74 和0.17 eV 2 个能垒(见图10(f)∼(h)). 这些能垒表明COOH* 和CHO* 的旋转过程在室温下会快速发生, 说明COOH* 和CHO 的旋转是打破耦合关系、加快电化学过程的关键. 这也表明, 提高反应温度将进一步提高电催化CO2RR 的效率, 这可能为热催化和电催化的结合打开了一条新的思路.

图10 机器学习驱动的CO2RR 催化剂的发现Fig.10 Machine learning-driven discovery of HEA electrocatalysts toward CO2RR
目前, 机器学习在高熵合金电催化剂领域除了上述应用外, 在其他电催化反应中也逐渐引起人们关注. 例如, Wang 等[88]开发了一类结构有序的新型PtRhFeNiCu HEAs, 将其作为乙醇氧化反应的电催化剂; Feng 等[89]通过可扩展的合成策略合成了平均直径为1.68 nm 的超小高熵合金纳米颗粒, 在0.5 M H2SO4溶液中,-0.05 V (vs. 可逆氢电极(reversible hydrogen electrode, RHE)) 下获得了28.3 A·mg-1的超高质量活性. 目前, 由于电催化材料具有多样性,还没有形成统一的选择方法, 并且单一描述符无法描述整个电催化性能, 因此通常将这些描述符相互组合并进行综合应用, 以获得电催化剂的卓越性能. 原子半径、原子序数、配位数等几何描述符, d 带中心及相关性质、价电子等电子描述符, 以及吸附能、电负性、电子亲和、电离能等描述符是目前比较常用的描述符. 这些描述符的组合对于开发高效的机器学习应用程序至关重要. 总的来说, 这些工作表明机器学习模型在加速电催化剂设计方面具有较强的竞争力, 具有不错的预测精度, 机器学习将促进高熵材料在催化剂领域的快速发展.
4 总结与展望
机器学习及其组合方法已经成功地应用于电催化剂设计, 成为一种用于发现新的电催化剂的强大工具, 同时从现有数据集中提取知识. 然而, 利用机器学习设计新型电催化剂的挑战依然存在.
首先, 机器学习应用程序的标准数据集的缺乏限制了其更广泛的适用性. 尽管快速发展的大数据挖掘技术有望从大型数据池中提取有用的信息和知识, 但数据的多样性限制了其使用范围. 目前, 电催化剂的发现和优化主要依靠经验, 还没有足够精炼的相关信息来指导机器学习工作. 尽管机器学习在许多与电催化相关的应用中取得了成功, 但其引导的催化剂设计仍处于初级阶段.
其次, 如何有效地从机器学习中提取物理化学性质也是一个巨大的挑战. 通常, 机器学习方法不包含决定属性的物理定律, 这导致模型内不确定的误差传播. 不同机器学习模型的交叉验证可以帮助减少这种不确定性. 然而, 这种交叉验证方案需要一个代表整个化学体系的样本进行探索, 这是非常困难的, 因此代表性样本至关重要. 此外, 计算数据和实验数据的相互验证也是保证物理性质正确获取的潜在方法. 由于模型尺寸和模拟范围的限制, 计算数据通常比较简化, 故复杂的反应条件使得实验数据往往隐含叠加规律. 理论和实验数据的整合对电催化剂的物理性质的认识和未来电催化剂的发展具有重要的指导意义.
最后, 电催化通常发生在固液界面, 机器学习对电催化真实环境的模拟非常有限. 除了催化材料外, 还有溶液、电解质和施加的电压参与反应. 因此, 溶剂化效应、电解液效应和外加电压引起的电催化剂极化效应、双电层的形成等都是需要考虑的关键因素. 许多因素的叠加使得反应体系特别复杂, 故对固液界面的理解仍然非常有限, 且因实验的限制, 还缺乏基于实验观察在分子水平上的认识. 将数据科学与理论和实验方法相结合, 可能会产生发现电催化剂的新方法. 为了增加材料数据的数量, 研究人员应该从高通量计算中获得理论指标, 以便产生智能方法. 总的来说, 基于实验值, 通过机器学习设计电催化剂仍处于初级阶段. 在未来, 如果实验值可以方便地通过数据库检索到, 且在每次实验之前使用机器学习建模, 则将大大节省实验的时间和费用.
本工作综述了机器学习与高熵催化材料的概念以及机器学习在电催化剂设计中的应用,详细分析了机器学习在电催化领域的应用. 机器学习是探索非贵金属或低载量贵金属催化剂的有效工具, 为HER、OER、ORR 和CO2RR 等反应的高熵电催化剂设计提供了一种高效的新方法. 然而, 标准数据集、标准方法和系统指导的缺乏, 限制了其适用性范围, 挑战仍然存在. 此外, 电催化输入描述符缺乏详细的指导, 使其应用遇到了障碍. 随着现代数据科学的发展, 机器学习无疑将在辅助设计中发挥越来越大的作用.
