Pd 基氧还原反应纳米合金电催化剂研究进展
2023-12-17刘丹叶胡振亚崔朋蕾
刘丹叶, 曾 庆, 胡振亚, 刘 卉,崔朋蕾, 陈 东, 杨 军
(1. 中国科学院过程工程研究所, 北京 100190;2. 中国科学院大学材料科学与光电技术学院, 北京 100049)
电化学相关研究起源很早, 从1780年Galvani 发现生物电开始, 到1800年Volta 利用铜片和锌片发明了世界上第一个可以连续供电的装置——伏打电堆, 此时电化学才初步为人们所认识. 到了19 世纪和20 世纪, 电化学进入了高速发展阶段. 在这一时期, Faraday 提出了法拉第电解当量定律, 揭示了电量与电极反应物重量之间的关系, 同时也创造了“电解质、阴极、阳极、离子” 等专业术语, 为电化学研究奠定了基础. 1889年, 德国物理学家Nernst 建立了以自己名字命名的Nernst 方程, 进一步将化学能和原电池电极电位联系起来, 并以此获得了1920年诺贝尔化学奖, 凸显了这一方程的重大意义.
随着电化学测量方法和测试技术的成熟, 人们意识到了电极反应动力学的重要性. 一个重点关注电极界面反应过程的交叉学科——电催化应运而生. 电催化概念最早由Kobosev 等[1]提出, 直到最近几十年才被深入研究. 一般认为电催化中的催化作用来源于修饰的电极表面或电解液中的添加剂. 它是一个相对概念, 因为所有电极均有一定的催化活性, 而抛开电极的催化又不能称之为电催化. 目前电催化研究主要集中在改变或修饰电极, 探究合适的电催化剂,使电极/电解质界面上的电荷转移加快, 从而加速反应进程.
一般而言, 地球上某种资源(如H2O 和N2) 的丰富性意味着其具有良好的稳定性, 通过传统方式将其转化并加以利用会消耗大量能量. 电催化在电压作用下, 可以大幅降低反应的活化能, 推动反应转化的进行. 电催化可以贯穿到C、H、O 和N 等多种元素的物质循环之中.以丰富的N2、CO2和H2O 等为原料, 通过电催化可以制得高附加值的H2、碳氢化合物、氧化物和氨等化学品, 且这些过程的产物或者伴随的能量转化可以应用到能源、环境以及化工生产等领域(见图1, 其中TM 为transition metal, 过渡金属)[2].

图1 电催化转化促进碳循环、氮循环和氢循环的闭合Fig.1 Electrocatalysis conversion promotes the closure of carbon, nitrogen, and hydrogen cycles
电催化通常涉及两个或两个以上反应步骤, 产生多种中间态物种. 根据反应机理不同,电催化可以分为两类反应: 在第一类反应中, 反应物离子或分子经过电子传递形成中间态物种并吸附于电极表面, 然后经过一系列反应生成稳定的分子, 析氢反应(hydrogen evolution reaction, HER)、氢氧化反应(hydrogen oxidation reaction, HOR) 以及氧还原反应(oxygen reduction reaction, ORR) 均属于这一类反应; 在第二类反应中, 反应物分子首先在电极表面进行吸附解离或者缔合, 然后再发生电子转移进行化学转化, 甲酸氧化反应(formic acid oxygen reaction, FAOR)、甲醇氧化反应(methanol oxygen reaction, MOR) 和乙醇氧化反应(ethanol oxygen reaction, EOR) 均属于这一类反应[3]. 通过了解电极反应机理, 可以指导设计相对应的电催化剂, 促进整个电催化过程向有利的方向进行. 本工作先以燃料电池阴极发生的ORR 过程为例, 介绍了电极反应过程涉及的反应机理.
1 ORR 电催化机制
ORR 作为各类燃料电池以及金属-空气电池的阴极反应, 其反应速率远低于阳极氧化反应, 反应的过电位也较高. 因此, 增强ORR 催化剂性能对于提升整个燃料电池的输出电压和能量效率极为关键. 根据反应过程中转移电子数的不同, ORR 可以分为四电子反应和二电子反应. 由于四电子反应的理论电压远高于相应的二电子反应, 因此计算反应过程中所转移的电子数是评价ORR 催化剂性能的重要指标之一.
ORR 的反应机理通常分为酸性和碱性两种情况[4-5]. 在酸性条件下, O2可转移4 个电子生成H2O, 也可转移2 个电子生成H2O2, 而H2O2有可能继续反应生成H2O, 具体过程如下,其中SHE 表示标准氢电极(standard hydrogen electrode):
ORR 的总电极反应较为简单,但其中包含多步基元反应.二电子反应中涉及HOO-、HO-等物种的生成和转化, O2中的O—O 键在逐步加氢过程中未断裂. 转化过程如图2[4]所示.

图2 酸性条件下ORR 的二电子反应路径Fig.2 Two-electron reaction pathway of ORR under acidic conditions
相比酸性条件, 碱性条件下ORR 的反应机理和理论电压均不相同, 但仍然是四电子反应优于二电子反应, 具体过程如下:
在这两种环境中, 二电子反应的能垒低于相应的四电子反应, 因此在大多数催化剂表面发生的是二电子反应, 少数催化剂表面可以发生四电子反应. 基于此, 设计合成能够遵循四电子反应的ORR 电催化剂已成为燃料电池领域的热点研究.
2 Pd 基纳米合金中的物理效应及其电催化ORR 研究进展
纳米材料几何尺寸较小、比表面大, 可以暴露更多可利用的表面原子, 具有较高的本征活性, 因而在催化剂相关的科学研究和工业应用方面受到广泛关注. 在具体催化体系中, 催化剂通常由纳米颗粒和载体(如活性炭、石墨烯等) 两部分组成. 催化剂为电催化反应提供活性位点, 可供反应物分子吸附、转化以及产物释放, 因此可以通过催化剂表面改性来优化其与特定分子的结合状况, 从而达到提升催化性能的目的. 电催化剂的活性主要取决于活性组分的本征活性和暴露的活性原子数量, 因此所有提升催化剂性能的策略都可以归结于这两方面的优化.
已有研究提出了一些联系催化剂活性与电子构型的理论和模型, 以期揭示不同电催化剂结构与活性的关系. Nørskov 等[6-9]提出的d 能带模型可以较好地描述金属催化剂的电子构型与吸附物种的吸附强度之间的关系, 因而被广泛应用于催化剂设计之中. 该模型认为, 吸附分子在金属表面的吸附和解离活化能大小, 与吸附物种能级和过渡金属d 能带耦合程度有很强的相关性. 因此, 可用d 轨道态密度中心εd(d 带中心) 作为吸附能的描述符, 通过εd与费米能级(Fermi level,εF) 之间的差值来确定金属表面与吸附物种之间的吸附强度[9].
吸附物种反键轨道的填充情况与其在催化剂表面的吸附密切相关, 而εd的位置可以影响反键轨道的状态. 当εd靠近费米能级时, 较多电子填入反键轨道, 吸附物种会与金属表面产生较强的相互作用; 反之, 当εd远离费米能级时, 则相互作用较弱. 对于特定的活性组分, 其εd通常是一定的, 但其周围化学环境的变化会引起εd的上升或下降, 进而达到调节金属表面活性位点反应活性的目的(见图3)[10]. 配体效应(或称为电子效应) 和应变效应都会引起εd的改变, 显著影响反应中间体在催化剂表面的吸附能, 从而可能改变反应路径和能垒(见图3(a)和(b)). 几何效应和协同效应(双功能机制) 也能改变催化剂的活性(见图3(c) 和(d)). 催化剂中各种物理效应往往同时存在, 共同影响反应进程.
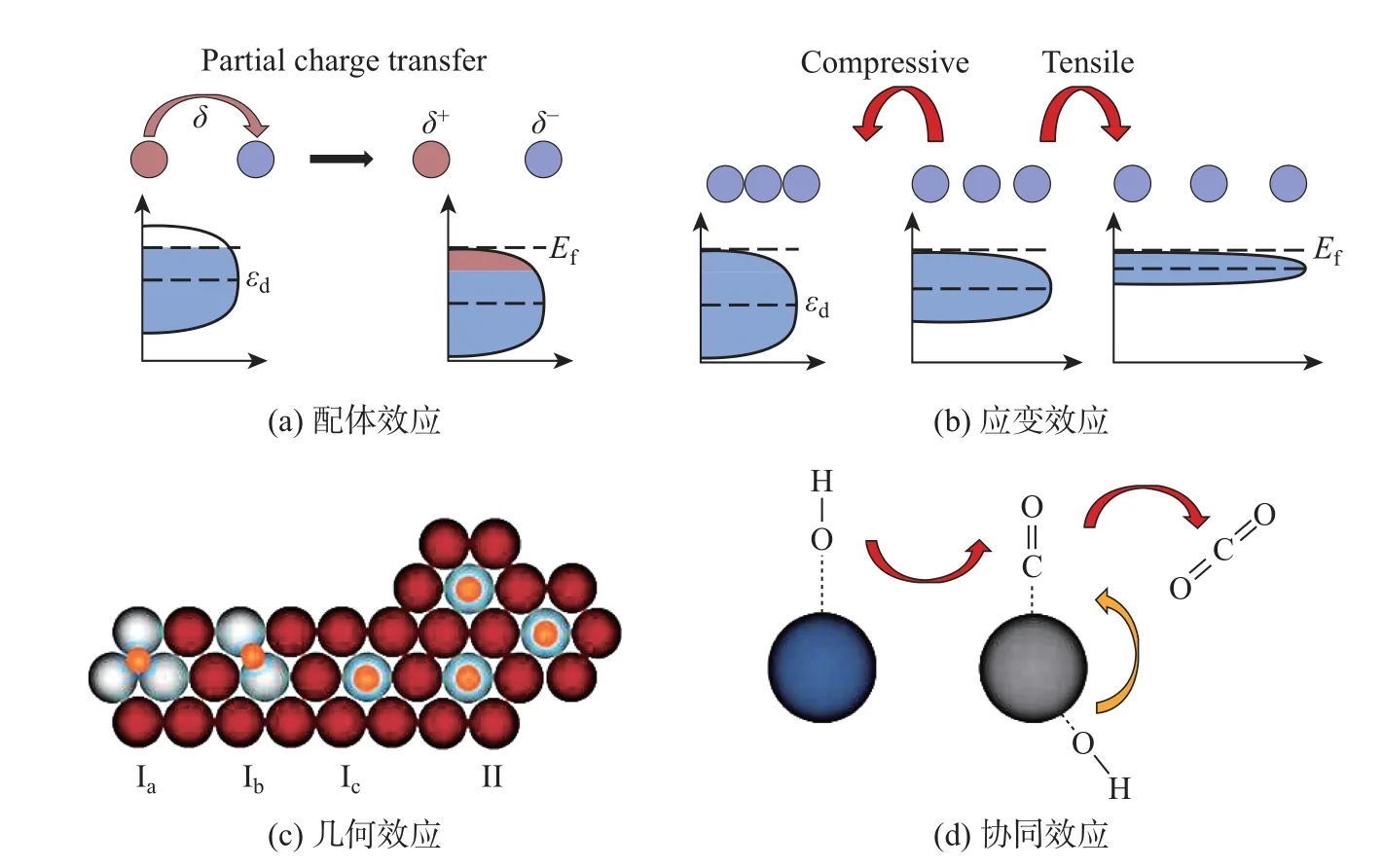
图3 合金电催化剂的物理效应Fig.3 Physical effects in alloy electrocatalysts
目前, 电催化剂研究的目标是在保证催化剂活性和稳定性的同时, 尽可能减少贵金属的用量. 因此, 电催化剂研发的关键在于解析和利用上述物理效应, 最大限度地发挥活性组分的催化作用. 本工作以综述Pd 基纳米合金为主, 但为了充分说明催化剂中的物理效应, 也会涉及一些Pt 基催化剂的研究进展和ORR 之外的电化学反应, 例如MOR 和EOR.
2.1 几何效应
在电催化剂研究早期, 人们比较关注催化剂的尺寸、形貌等空间几何结构. 对于催化剂来说, 只有表面一小部分原子参与反应过程, 内部大部分原子无法接触到反应物, 因而未得到充分利用. 当颗粒尺寸减小时, 其表面积呈指数增加, 表面原子所占的比值快速增大, 使得同样组成的催化剂的原子利用率得到提高[11]. 另外, 小颗粒的表面能较高, 很容易与反应物结合,反应活性更高. 从工程角度分析, 利用催化剂的尺寸效应可以直接降低成本, 对于需要使用较高成本贵金属催化剂的反应更有意义; 从基础科学角度分析, 调节催化剂的尺寸也会改变催化剂和相应表面的本征活性, 对探究催化活性来源有一定意义(见图4)[12].
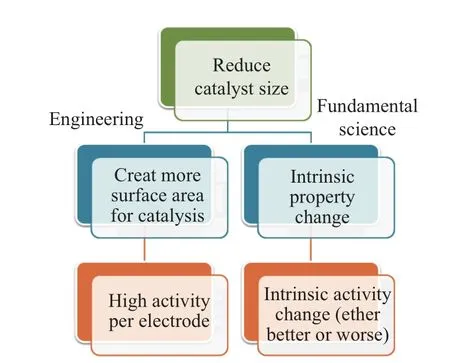
图4 工程和基础科学角度下的催化剂尺寸效应Fig.4 Size effects of catalysts from the aspects of engineering and fundamental science
Jiang 等[13]在N2/H2气氛、300∼600◦C 条件下, 对负载量为20% (质量比) 的Pd/C 催化剂进行加热处理, 得到粒径为3.0∼16.7 nm 的Pd/C 催化剂. 该研究将不同粒径的Pd/C 催化剂应用于催化ORR 过程, 探究了ORR 对Pd 催化剂的尺寸依赖性, 结果如图5 所示. 可以看出: 当电压为0 V vs. Hg/HgO/OH-时, 面积活性(specific activity, SA) 和质量活性(mass activity, MA) 与Pd 粒径之间的关系并不一致; 当颗粒较大时, Pd/C 催化剂的面积活性较大;随着Pd 粒径减小, Pd/C 催化剂的质量活性呈现先增大后减小的趋势; 当Pd 粒径为5 nm 左右时, Pd/C 催化剂的质量活性最大.

图5 Pd/C 催化剂的粒径与ORR 质量活性和面积活性的关系Fig.5 Dependence of mass activity and specific activity for the ORR on Pd/C catalysts with different Pd particle sizes
Zhou 等[14]在水相中合成了负载型Pd 纳米催化剂. 该研究通过调整柠檬酸钠与Pd 前驱体的比例改变Pd 粒径, 直接得到了粒径在2.7∼8.7 nm 之间的Pd 纳米颗粒. 电化学测试结果显示, 室温下对ORR 最有利的Pd 粒径也在5.0∼6.0 nm 之间. Shim 等[15]探究了Pd 纳米颗粒在更小粒径范围(0.5∼3.0 nm) 内在酸性电解质溶液中催化ORR 时的尺寸影响. 该研究发现, 粒径较小的Pd 表现出了更正的ORR 起始电位(或半波电位), 且在ORR 过程中转移了更多电子, 具有更好的电催化性能. 与Pd 类似, Shao 等[16]研究了ORR 对Pt 颗粒尺寸的依赖性. 在1∼5 nm 粒径范围内, Pt 纳米颗粒对ORR 的催化活性随着粒径的减小呈现先升高后降低的趋势, 存在一个最佳催化尺寸. 该观点与Mayrhofer 等[17]的研究结果相吻合, 但是对于最佳催化尺寸的具体数值在已有研究中并未达成一致. 这可能是由于催化剂合成方法和电化学测试条件不同而导致的[16-20]. 除了球形颗粒, 其他形貌的催化剂也可表现出一定程度的尺寸依赖, 比如Koenigsmann 等[21]发现Pd 纳米线长度也会对其ORR 催化性能产生影响.该研究通过比较不同长度(270、45 和2 nm) Pd 纳米线的ORR 性能, 发现催化时具有明显的尺寸效应, 即随着纳米线长度的减小, 电催化活性提高了近2 倍.
作为尺寸降低的极限, 早在1995年, Thomas 等[22]就首次提出了构建孤立原子作为异质催化剂活性位点的设计概念. 该研究将有机金属配合物直接接枝到介孔二氧化硅MCM-41 的内壁上, 产生了一种形状选择性催化剂. 该催化剂具有大量可接近的、间隔良好且结构明确的活性位点, 可用于催化环烯烃的环氧化反应, 但是并没有直观的证据证实孤立原子的存在. 近几年, 通过将先进的表征手段与新的合成方法相结合, 使得单原子研究快速发展, 已成为电催化领域比较热门的研究方向. Qiao 等[23]在报告中指出, 分散在FeOx上的单个Pt 原子具有高度稳定性. 这项工作使用了高角度环形暗场模式下扫描透射电子显微镜(high angle annular dark field-scanning transmission electron microscope, HAADF-STEM), 结合光谱和理论计算技术阐明了单个Pt 原子与FeOx载体之间的键合和催化机制, 并且首次提出了单原子催化剂(single-atom catalysts, SACs) 的概念. 研究表明, SACs 提高各种反应催化活性的原因可以归结于活性中心独特的电子结构和不饱和配位环境. 该类型催化剂利用各种配体将单个原子固定在氧化物或石墨烯等载体表面, 可以使催化过程中活性位点和反应机理的研究更加方便[23-27]. Xiang 等[28]制备了一种3D 交织结构, 将Pd 原子沉积在均匀结合的MnO2纳米线和碳纳米管(Pd/MnO2-carbon nanotube, Pd/MnO2-CNT) 表面, 耦合了金属氧化物中Pd 原子的优越催化性能与CNT 的高导电性(见图6). 这种纳米复合材料可以稳定地催化ORR 和析氧反应(oxygen evolution reaction, OER), 其中Pd/MnO2-CNT 在催化ORR 过程中表现出了较大的质量活性, 比Pd/CNT 高2 个量级, 比现有的Pd 基催化剂高5 倍. 通过实验和理论研究发现, Pd 和周围金属位点的协同作用是大大加速ORR 反应动力学的原因. 该作用激活了氧气分子, 优化了Pd 原子与反应中间产物的结合强度.
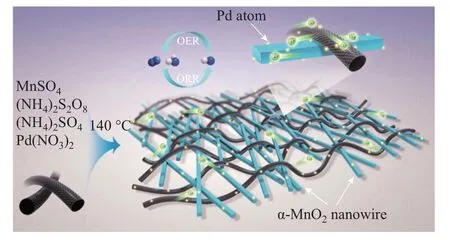
图6 3D 交织CNT 和MnO2 纳米线锚定Pd 原子用于双功能OER 和ORR 电催化Fig.6 3D interwoven CNT and MnO2 nanowire anchored with Pd atoms for bifunctional OER and ORR electrocatalysis
除了单金属, 双金属中的尺寸效应同样不容忽视. 由于双金属中不同粒径颗粒的化学组成难以完全保持恒定, 使得单一研究尺寸效应对催化活性的影响有一定困难. 在已有研究中,Zhang 等[29]通过在不同温度下热处理PdCo 纳米颗粒, 得到了不同粒径的PdCo 纳米颗粒(见图7). 可以看到, 当处理温度为300◦C 时, PdCo 纳米颗粒的平均粒径增大到8.9 nm, 其催化ORR 的活性明显增强. 但是催化活性的增强并不能完全归因于粒径的改变, 热处理过程可能会使催化剂的表面发生改变, 影响其催化性能. 此外, Gan 等[30]通过简单调节1,2-十四碳二醇的加入量, 得到了不同尺寸(3∼10 nm) 且组成恒定的PtNi3纳米颗粒, 并探究了该颗粒的尺寸效应对ORR 催化性能的影响. 结果表明, 6∼8 nm 粒径范围内的PtNi3纳米颗粒表现出了最高的ORR 催化活性和稳定性, 为双金属ORR 电催化剂的尺寸选择提供了重要的依据.

图7 热处理温度对催化剂(Pd-Co/C 合金) 粒径的影响Fig.7 Effect of heat-treatment temperature on the catalyst (Pd-Co/C alloy) particle size
一般而言, 金属催化剂大多具有排列规则的晶体结构, 由多种可以标记为(hkl) 的晶面组成. 在所有晶面中, (111)、(100) 和(110) 这3 个晶面表面原子的配位数分别为9、8 和7, 性质较为稳定, 属于低指数晶面. 当h、k 和l 中至少一个指数大于1 时, 所形成的晶面为高指数晶面. 高指数晶面通常含有较多台阶原子和扭结原子. 这些原子表面配位数低, 因而具有较高的催化活性. 另外, 随着制备方法逐渐多样化, 具有特殊形貌结构的催化剂被广泛报道, 如纳米片、纳米线、立方体、多孔结构以及框架结构等. 特殊形貌催化剂具有如下优势: ①这些催化剂的形成可能是由于某些高指数晶面的暴露, 而高指数晶面具有较高的化学反应性; ②特殊结构如纳米片、多孔结构等有利于反应过程中物质扩散和传递, 加快反应速度; ③同样质量的同种催化剂, 特殊形貌催化剂可利用的活性原子数目较多, 使得整体催化性能提高.
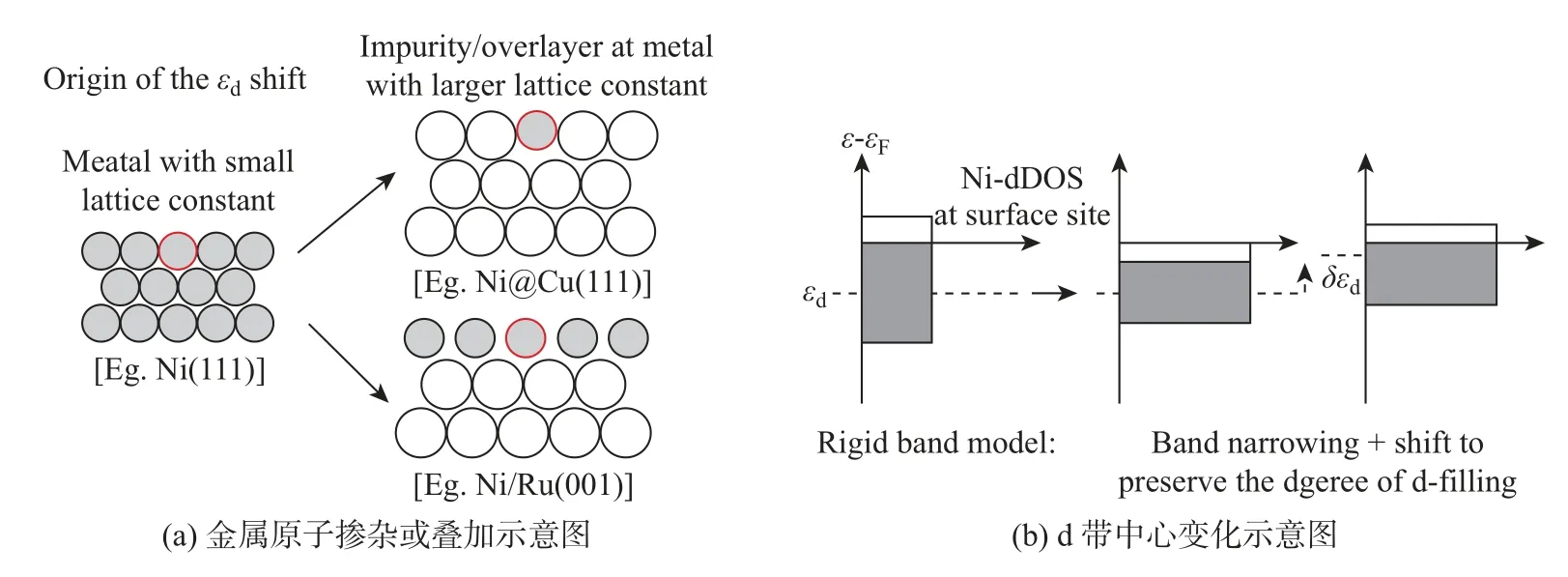
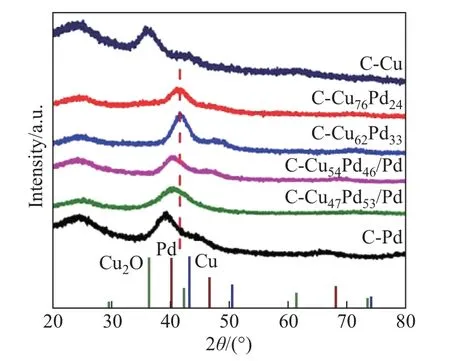
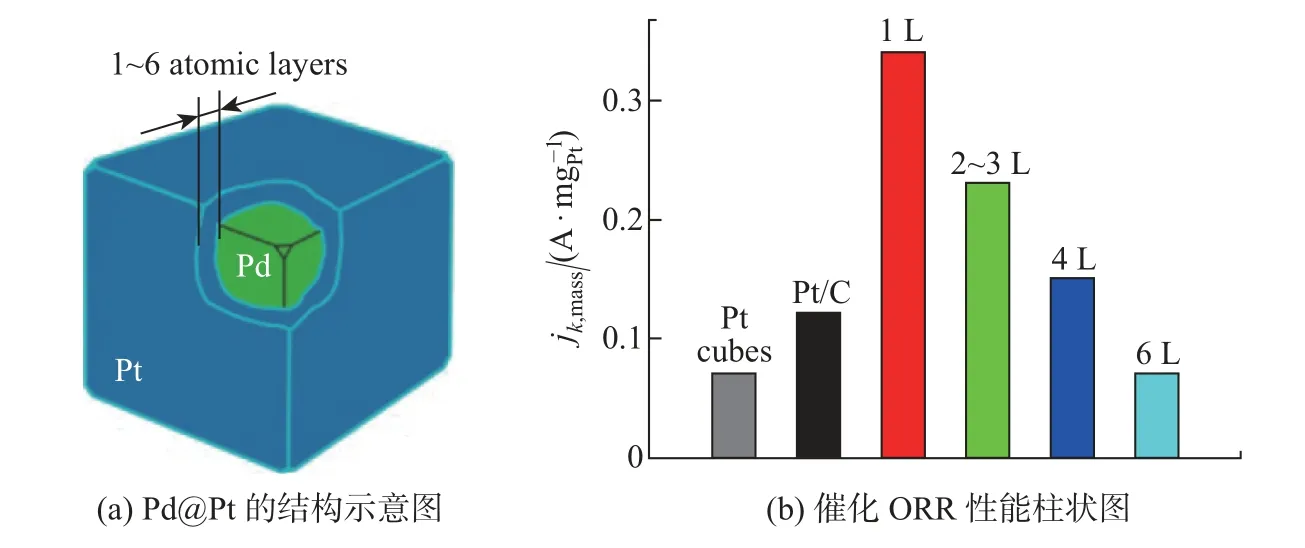
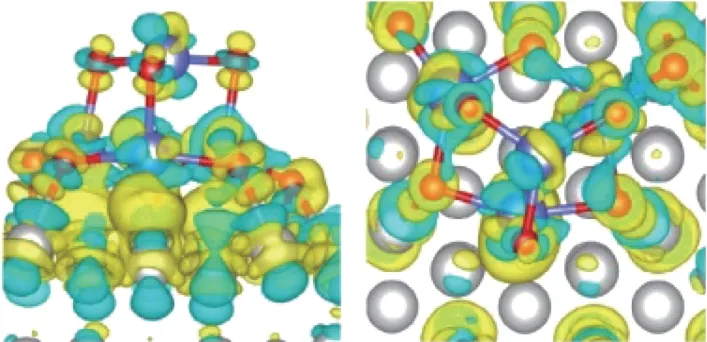
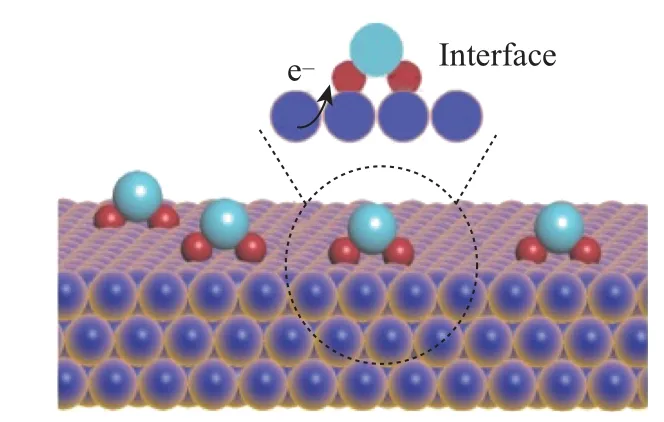
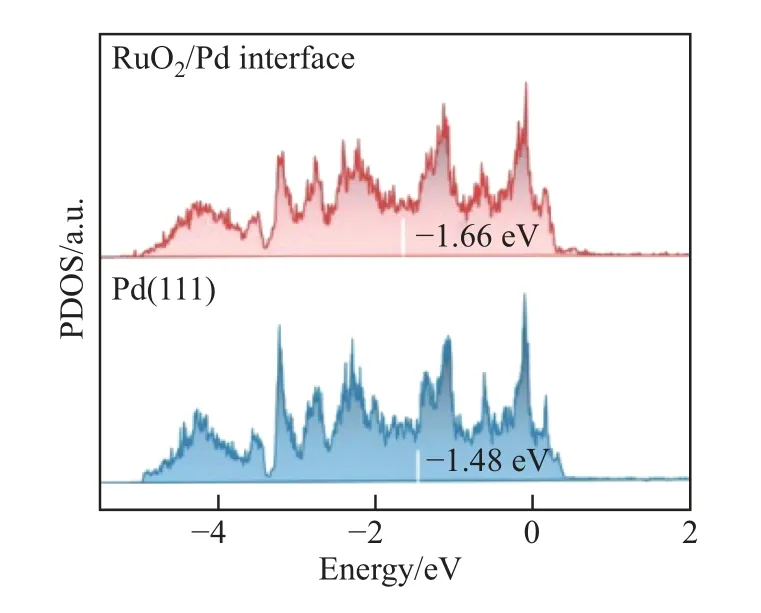
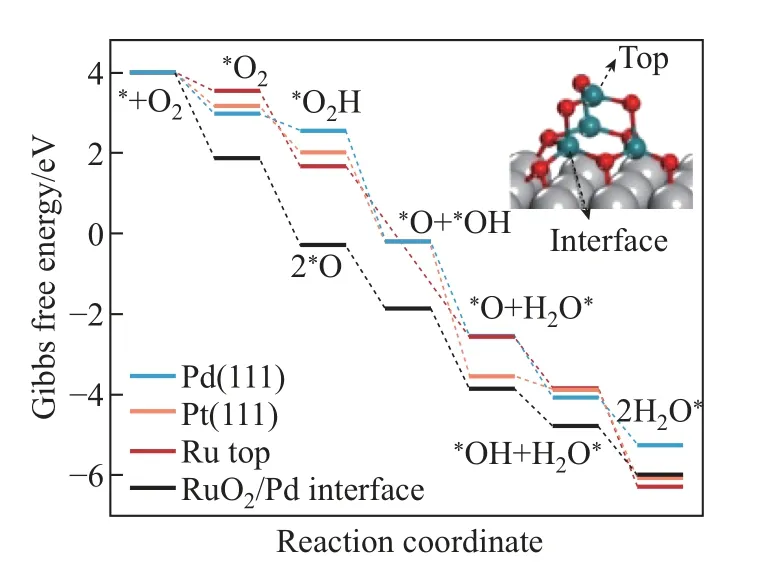
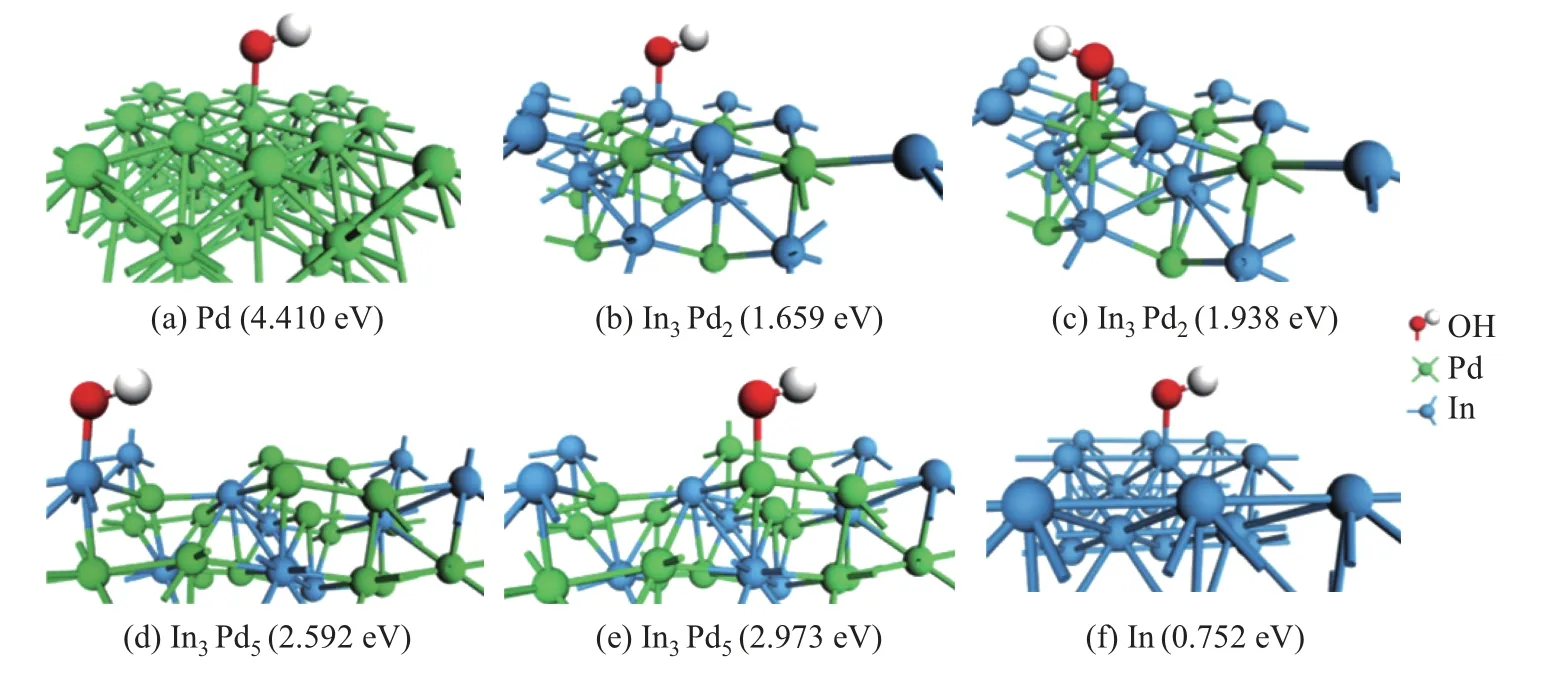
研究者利用各种表面保护剂或者辅助剂定向诱导合成由特定晶面组成的催化剂. Kiguchi等[31]研究了在不同阳离子的碱性溶液中, 不同低指数Pd 晶面的ORR 催化活性. 该研究合成了粒径约为3 nm 并且暴露(110)、(111) 和(100) 的Pd 单晶结构. 电化学性能测试结果显示, 在不同的碱性电解质中, ORR 催化活性顺序均为Pd(110) 具有低配位数的高指数晶面往往具有更高的催化活性, 但是暴露高指数晶面催化剂的大量制备仍然是一个挑战. 例如, Wang 等[32]报道了具有低配位的Pd 截面四面体的合成, 并作为碱性条件下ORR 电催化剂, 其面积活性/质量活性(SA/MA) 为2.46 (mA·cm-2)/1.69, 分别是商业Pd/C 和Pt/C 的12.3/16.9 和10.7/14.1 倍. Qi 等[33]采用温和可控的种子介导生长方法合成了具有高催化活性的Pd 多孔单晶纳米花(porous single crystal nanoflowers, PSNFs). 该催化剂暴露出具有较高催化活性的(100) 晶面和较多的台阶原子. 该Pd PSNFs 的形成机制如图8 所示. 在反应开始的20 min 内, Pd 纳米颗粒为立方体形状, 但尺寸迅速增大; 一段时间后, 每个纳米立方体的6 个面都形成了一个微小的凹面; 随后, 凹面纳米立方体转变为凸面纳米立方体; 最后, 转变为球形花朵状, 并且尺寸逐渐增大. 研究认为影响催化剂结构的因素有3 个速率: Pd 前驱体向纳米立方体种子的扩散速率(V1)、Pd 原子的生成速率(V2) 和Pd 前驱体在纳米立方体表面的消耗速率(V3). 由于其特殊结构, Pd PSNFs具有较强的乙醇氧化反应活性、较高的抗CO 中毒性和稳定性. 这些都远远优于Pd 纳米立方体, 以及以(111) 面为主的树突状海胆样Pd 纳米颗粒和Pd/C 催化剂. 图8 Pd PSNFs 的生长机制Fig.8 Growth mechanism of the Pd PSNFs 特殊形貌结构的催化剂可以为电催化反应过程提供更大的活性表面和更多的活性位点.金属烯与纳米片相比只有原子厚度, 表面有丰富的金属活性位点, 原子利用率高、电导率高,在电催化反应方面具有很大的潜力. Yu 等[34]将富缺陷的超薄多孔Pd 金属烯催化剂应用于ORR 反应, 发现其具有优异的催化活性和稳定性. 由图9 所示的HAADF-STEM、透射电子显微镜(transmission electron microscope, TEM)、原子力显微镜(atomic force microscope,AFM)、高分辨率TEM (high resolution TEM, HRTEM) 观测结果所示, 得益于其特殊的薄片和多孔结构, Pd 金属烯的电化学活性面积为66.7 m2/g, 高于Pd/C、Pt/C 和Pd 纳米片(nanosheets, NSs)/C 的活性面积. Pd 金属烯在空间中会发生弯曲, 引起应变效应, 改变Pd 的电子结构, 优化Pd 金属烯表面的氧分子吸附. 此外, Pd 金属烯具有较多Pd 缺陷. 这些缺陷能够为O2的吸附提供活性位点,促进O2的活化. 综合该结构的多方面优势,在0.1 mol/L KOH电解液中,0.9 V 时Pd 金属烯的ORR 质量活性为0.892 A/mgPd,面积活性为1.336 mA/cm2,分别是商业Pt/C 催化剂活性的5.1 倍和4.0 倍. 除了超高的活性, 经过5 000 圈加速容忍性测试后, Pd 金属烯的ORR 极化曲线基本与初始曲线重合, 表明其具有良好的稳定性. 图9 Pd 金属烯的HAADF-STEM 图像、TEM 图像及对应的选区电子衍射模式、AFM 图像及对应的高度剖面和HRTEM 图像Fig.9 HAADF-STEM images, TEM images and corresponding selected area electron diffraction,AFM image and corresponding height profile, and HRTEM image of Pd metallene 向Pd 中引入其他组分后, 同样可以形成具有特殊形貌的催化剂. 但即使引入同一组分,在不同表面活性剂作用下也会生成不同结构的催化剂. L¨u等[35]报道了一种利用二十八烷基二甲基氯化铵的两亲性合成双金属PdAg 单晶纳米线(single crystal nanowires, sNWs) 的方法.动力学研究表明, 超薄PdAg sNWs 的协同电子效应和特殊结构效应显著提高了其乙醇氧化的催化性能. Huang 等[36]则通过以十八烷基三甲基氯化铵为结构导向剂, 在水溶液中共还原Pd 和Ag 的前驱体, 得到了厚度为5∼7 nm 的2D AgPd 纳米片, 其特殊结构同样在碱性溶液中表现出了显著增强的乙醇氧化电催化活性和稳定性. Zhang 等[37]首次合成了Pd-Sb 纳米片, 可用于同时促进阴极ORR 和阳极乙醇氧化燃料电池反应. 介孔结构与纳米片、纳米线结构类似, 具有较大的活性表面积. 此外, 丰富的孔道结构有利于加快反应过程中的物质传递. Iqbal 等[38]的研究表明, 可以通过嵌段共聚物的水诱导胶束化得到PdCu 介孔纳米微球, 其多孔性使颗粒内外表面的大量活性位点参与到反应中, 有利于ORR 过程中的物质传递. 与之类似, Deng 等[39]报道了一种简便合成三金属PtPdRh 介孔纳米球(mesoporous nanospheres, MNs) 的方法. 研究发现, 引入合适浓度的表面活性剂F127,以及精确控制各种金属前驱体的还原动力学是提高收率的关键. 合成的PtPdRh MNs 具有比表面积大、质量扩散良好、电催化剂利用率高等优点. 此外, 得益于多金属组成和独特的协同效应, PtPdRh MNs 的ORR 电催化性能明显优于双金属PtPd MNs 和商业Pt/C 催化剂. 晶格应变定义为颗粒表面或局部原子与原子之间距离的改变, 其中比纯金属标准原子间距偏大的称为晶格拉伸效应, 偏小的称为晶格压缩效应[40]. 表面原子配位数改变和偶然出现的无序原子都会引起局部晶格应变, 因此应变是固体材料中的固有性质[40-42]. 对于纳米结构材料, 引入更加显著的晶格应变是一种优化其能带结构, 达到提升催化性能的非常有效和经济的方法. 根据d 轨道中心理论, 金属的配位数变化可以导致其d 带中心发生变化. 如图10 所示(其中DOS 为density of state, 即态密度; dDOS 为d 型DOS): 当一个晶格常数较小的金属原子灰色小圆(如d 带超过一半电子填充的过渡金属Ni) 作为杂质或叠加在一个晶格常数较大的金属原子白色大圆(如Cu 或Ru 晶格) 表面时, 会引起晶格拉伸; 拉伸应变会降低配位数, 从而导致嵌入密度降低、d 带宽度变窄, d 带中心相对于费米能级发生上移, 以保持d 轨道的电子填充(灰色阴影区); 随着d 带中心的上移, 金属表面与反应中间体的吸附作用得到增强; 反之, 压缩应变具有相反的作用, 导致d 带宽度变宽和d 带中心下移, 最终产生较弱的吸附作用[7]. 图10 具有较小晶格参数的金属原子在极大晶格参数表面掺杂或叠加时相应的d 带中心变化示意图Fig.10 Schematic illustration showing the put of a metal atom with a small lattice constant as an impurity or overlayer at the surface of a metal with a large lattice constant and the corresponding changes in d band center 在纳米材料中, 应变不仅改变了原子构型, 而且改变了电子轨道的重叠、电荷分布和键合强度, 还可能会导致新的物理和化学性质, 如电子、光学、磁性、声子和催化性质等[43].目前, 可以通过多种技术来直接表征表面应变效应, 例如球差校正HAADF-STEM[44]、X-射线衍射(X-ray diffraction, XRD)[45]、X 射线光电子能谱(X-ray photoelectron spectroscope,XPS)[46]、广角X-射线散射(wide angle X-ray scattering, WAXS)[47]、X-射线吸收光谱(X-ray absorption spectroscope,XAS)[48]和X 射线发射光谱(X-ray emission spectroscope,XES)[49].已知晶格错配、颗粒尺寸、结构缺陷和形貌等多种因素都可以引起应变. 如图11 所示, 根据结构的不同, 可以将已报道催化剂中的晶格应变分为5 种类型, 即核壳结构催化剂、负载型颗粒/团簇催化剂、缺陷、尺寸/形状变化和掺杂固体合金[40], 其中核壳结构中核与壳之间由于晶格错配引起的晶格应变最为显著和典型. 衡量应变强度参数ε的计算公式为 图11 不同类型的应变形成机制Fig.11 Formation mechanisms and types of different strains 式中:dp表示颗粒表面或局部区域的原子间距;ds表示相应纯金属材料标准原子间距[50]. 常用来构建核壳结构的方法有种子生长法、电化学置换反应法、去合金法和热退火诱导偏析法等[49,51-53]. 应变效应调节催化剂反应活性的策略对于多个电极反应过程都是行之有效的. 例如, 通过对ORR 机理和转化过程研究发现, 纯Pt 或Pd 催化剂对于O、OOH 等中间物种的吸附太强, 限制了ORR 的速率. 通过向Pt 或Pd 中引入晶格常数较小的过渡金属, 诱导产生晶格压缩应变效应, 会减弱Pt 或Pd 表面与O、OOH 等物种的结合能力, 促进ORR 进行[54]. Kuttiyiel 等[55]研究了核壳结构中不同内核对催化活性的影响. 该研究以IrM (M=Fe、Co、Ni 或Cu) 合金作为内核, 通过电化学置换的方法, 在表面诱导生成Pt 单原子层(monoatomic layer, ML). 结果表明, IrM 内核能诱导更强的晶格应变, 这使得IrM 可作为Pt ML 电催化ORR 的较好衬底. Gan 等[56]合成了一系列具有不同组成的PtNi 合金(PtNi、PtNi3和PtNi5) 纳米颗粒(nanoparticles, NPs), 然后通过电化学去合金的方式脱除Ni, 制备得到不同Ni 含量的核壳型Pt-Ni (D-PtNi) 纳米材料, 其中在D-PtNi3催化剂上, Ni的富集会导致Pt 表面产生较大程度的压缩应变, 从而使催化剂的ORR 活性达到最大值. Koh等[57]同样利用电化学去合金的方法处理CuPt 合金, 得到了由纯Pt 包覆在CuPt 内核表面的CuPt@Pt 结构催化剂.该研究认为,去合金化处理使得颗粒表面Pt 原子的结构排布发生变化,Pt 原子间距改变, 更有利于ORR 的进行. Xie 等[58]和Jiang 等[59]合成了PdCu/Pd 核壳结构催化剂(见图12, 其中红球和白球分别代表Cu 和Pd 原子; TDPA 为tetradecylphosphonic acid, 即四癸基膦酸; TOA 为trioctylamine, 即三辛胺), 具有大小均匀、形态规则的特点(见图13). 图14 显示的XRD 图谱表明CuPd/Pd 纳米颗粒的衍射峰向纯Pd 的方向偏移, 在一定程度上证实了富Pd 表面的生成. 通过密度泛函理论(density functional theory, DFT) 计算表明, CuPd 合金核诱导0.5 nm 厚度的Pd 壳层产生压缩应变, 导致Pd 的d 带中心下移,减弱了*H 与含氧中间体在催化剂表面的结合强度, 促进了反应动力学, 显著提升了其催化HER/ORR 的活性(见图15). 图12 Cu-Pd 合金和CuPd/Pd 核/壳纳米颗粒的形成机制Fig.12 Formation mechanism of Cu-Pd alloy and CuPd/Pd core/shell NPs 图14 C-Cu、C-Cu-Pd、C-Pd 样品的XRD图谱Fig.14 XRD patterns of C-Cu, C-Cu-Pd,and C-Pd samples 图15 Cu54Pd46/Pd 纳米颗粒的HRTEM图像Fig.15 HRTEM image of Cu54Pd46/Pd NPs 值得注意的是, 壳层的厚度与晶格应变程度密切相关, 自然也影响着催化剂的活性. Xie等[60]合成了大小均匀的核壳型Pd@Pt 纳米立方体, 电镜表征和能量色散X 射线能谱(energy dispersive X-ray spectroscopy, EDX) 线扫描分析都可证实Pd 表面覆盖了Pt 原子层. 该研究通过调整Pt 前驱体的注入速度和反应温度, 可以精确控制Pt 原子的层数. 将合成的Pd@Pt纳米立方体用于ORR 电催化中时发现: 当Pt 在Pd 表面沉积1 层时, 质量活性最高(见图16); 沉积2∼3 层时, 面积活性最高. 类似地, Wang 等[61]同样在二十面体Pd 的表面沉积超薄Pt 壳层, 并由DFT 计算得到, Pd 内核压缩了表面Pt 的晶格, 减弱了其表面对OH 的吸附,使得其ORR 的面积活性和质量活性分别比商业Pt/C 催化剂提高了8 倍和7 倍. 除此之外,还有一些关于其他类型晶格应变对ORR 过程影响的研究. 例如, Sneed 等[62]合成了两种不同金属相的网格状排列Pd-Rh 纳米盒结构. 结果表明, 表面金属相纳米级分离产生的晶格应变起着重要作用, 影响着其在ORR 反应中的催化行为. 图16 Pd@Pt 的结构示意图及催化ORR 性能柱状图Fig.16 Structure diagram and ORR catalytic performance diagram of Pd@Pt 综上可知, 在表面应变工程中, 构建核壳结构是一种提升催化剂本征活性的重要手段. 此外, 若中间内核使用廉价金属还可以降低催化剂的成本. 但一般来说贵金属原子更稳定, 因此与其他金属在同一体系中时更易还原, 多分布于内核. Wang 等[63]利用DFT 计算研究了一些二元合金纳米颗粒系统(元素周期表中的第8∼11 周期), 探索了它们的偏析能, 确定了金属形成核-壳结构的倾向, 具体结果如图17 所示, 其中左边(上) 的金属与右边(下) 的金属合金时,几乎总是倾向于形成内核. 图17 12 种后过渡金属组成的55 个纳米颗粒中杂质偏析能DFT-PW91 的颜色编码矩阵Fig.17 Color-coded matrix of DFT-PW91 segregation energies for impurity in 55-atom NPs composed of 12 late-transition metals 在电催化反应中, 催化剂与表面吸附物质之间相互作用的本质是电子转移, 因此催化剂中活性组分的电子结构会影响反应历程. 配体效应又称为电子效应, 是指两种不同化学性质的原子形成合金或异质结构时, 电子的再分配和电荷转移现象[64]. 这种电子相互作用同样会引起电子结构(或d 轨道态密度中心) 的改变, 从而影响催化剂的表面反应活性. 一般来说, 只要将两种不同原子相接触, 就会产生或强或弱的配体效应. 但是这种表面配体效应是一种短程相互作用, 仅在催化剂表面影响较大, 若掺杂的原子位于纳米材料内部, 表面配体效应则可以忽略不计[65]. 与配体效应发挥作用的范围(1∼2 个单原子层) 相比, 晶格错配产生的应变效应则为长程效应(通常距离表面小于6 个单原子层), 其改变吸附物质吸附能的能力更强. L¨u等[66]巧妙利用一锅法在2D 超薄Pd 纳米片表面原位生成原子分散的岛式RuOx. 该催化剂对ORR 表现出了良好的催化活性, 其质量活性分别达到了商业Pd/C 和Pt/C 催化剂的22.4 和8.0 倍. 该研究通过∆ρ=ρTol-ρRuO2-ρPd(111)计算了RuO2覆盖Pd(111)切面的电荷密度差(见图18, 其中黄色和绿色区域分别代表电子的积累和耗散), 并结合投射态密度(projected density of state, PDOS) 研究了RuOx/Pd 界面的电子特性. 结果表明,电子有从Pd(111) 表面转移到RuOx/Pd 界面的趋势, 说明RuOx/Pd 界面处聚集了更多的电子(见图19). 图20 为RuOx/Pd 界面和纯Pd(111) 上Pd 原子的PDOS 图. 可以看到,RuOx/Pd 界面中Pd 原子d 带中心对应的数值为-1.66 eV, 低于纯Pd(111) 表面d 带中心的数值(-1.48 eV). 图21 为RuO2/Pd(111) 表面、纯Pd(111) 以及Pt(111) 表面不同催化位点上基本步骤的ORR 机理和相关的吉布斯自由能. 图22 为优化ORR 中间体在RuO2/Pd界面上的吸附构型, 其中灰色、灰绿色、红色和青色小球分别代表Pd、Ru、O、H 原子. 对RuOx/Pd 表面处ORR 催化过程进行理论计算, 发现其表面O 物种的吸附能降低. 这是由于其d 带中心下移导致. 这项研究证实了RuOx/Pd 界面存在的电子相互作用有利于吸附的O2进行活化、O—O 键的断裂以及中间含氧物种的还原和去除, 提高了整个ORR 效率. 除了Ru元素, Cu、Se 等与Pd 的电子相互作用, 对Pd 的ORR 活性也有类似的积极影响[67-68]. 图18 RuO2 覆盖Pd(111) 切面的电荷密度差Fig.18 Charge density difference of RuO2 coverage on Pd(111) facets 图19 RuO2/Pd(111) 界面上的电子转移模型Fig.19 Electron transfer model on RuO2/Pd(111) interface 图20 RuO2/Pd(111) 界面周围和纯Pd(111) 表面Pd 的d 带PDOS 图Fig.20 PDOS plots of d-band of Pd surround at the RuO2/Pd(111) interface and on pure Pd(111) surface 图21 RuO2/Pd(111) 表面、纯Pd(111) 以及Pt(111) 表面不同催化位点上基本步骤的ORR 机理和相关的吉布斯自由能Fig.21 Proposed ORR mechanism and relevant Gibbs free energy of elementary steps at different catalytic sites on the RuO2/Pd(111) surface and the pure Pd(111) and Pt(111) surface 图22 优化ORR 中间体在RuO2/Pd 界面上的吸附构型Fig.22 Optimized adsorption configurations of ORR intermediates on the RuO2/Pd interface 协同效应是一个广义概念, 同种成分承担反应不同步骤的活性位点, 或者其他组分(另一种原子、特殊载体和活性金属氧化物) 与主要活性组分共同完成整个反应过程的现象, 都可称为协同效应. 对于不同组分间协同效应的研究较为深入, 与单一组分相比, 多组分催化剂各个成分与反应物间的作用力强弱不同. 该研究同时进行特定物种的吸附和转化, 相互配合完成催化反应. 以双组分催化剂为例, 协同效应的具体反应方程可以表示为 式中: M1和M2分别代表两种金属; Aads和Bads分别代表两种表面吸附物种. 电催化中的协同效应最早由Watanabe 等[69]提出, 即在甲醇氧化过程中, 表面Pt 与Ru 原子相互配合共同发挥作用, 即所谓的双功能机理. 随后Yajima 等[70]采用衰减全反射技术(attenuated total reflection, ATR) 和原位傅里叶变换红外光谱(Fourier transform infrared spectroscopy, FTIR) 技术研究了溅射Pt-Ru、Ru和Pt 电极上MOR 的反应机理. 结果证实, 合金表面Pt 位点主要脱氢生成CO, 而Ru 位点则优先吸附水分子分解产生CO 氧化所需的氧. 这些结果支持了Pt-Ru 合金氧化甲醇的双功能机理. Lu 等[71]以表面可控PtNi 合金纳米颗粒为模型催化剂, 研究了其表面MOR 的性能和反应机理. 与Ru 的作用机制相类似, 在PtNi 催化剂中, Pt 提供CH3OH 吸附位点供CH3OH 转化为COads, 而Ni 提供OH 吸附位点. 当Pt-Ni 原子比为1∶1 时, MOR 的活性最佳. 这表明可提供CO 和OH 吸附位点的双官能团催化剂有望成为高活性MOR 催化剂. 在碱性条件下, Ni 对于Pd 表面催化MOR 过程同样具有极大的促进作用. Araujo 等[72]结合理论计算和实验验证, 更为深入地探究了Ni 增强MOR 性能的来源. 首先, Ni 加入后改变了MOR 的反应路线, PdNi 合金表面可以促进MOR 通过非CO 路线进行, OHads可以更容易地与CH2O 和CHO 反应, 开辟了两条不受CO 影响的生成CO2的路径. 其次, CO 脱附实验结果表明, PdNi 对CO 的氧化去除效率高于纯Pd 催化剂. 最后, Ni 的亲氧特性使其在MOR过程中更容易产生高度氧化的中间体. 这些因素均对MOR 过程有促进作用. 由于EOR 与MOR 类似, OH 物种与CO 中间物种反应, 完成氧化过程, 因此, 引入具有强吸附OH 物种的组分, 可以提高催化剂对EOR 的性能. 除了研究较多的Ru 元素以外, 还有一些其他的亲氧金属(Co、Ni、Sn、In 和Zn 等[73]) 以及氧化物表面有利于OH 的吸附, 可以促进紧紧吸附在Pt 或Pd 表面的CO 进一步转化. Chen 等[74]合成了单分散有序的In-Pd纳米颗粒作为新型高效催化EOR 的双金属催化剂, 其较高的活性可由两方面引发. 首先,In-Pd 纳米颗粒具有良好的CO 抗毒能力, CO 脱附实验证实了这一点. 其次, 通过对金属吸附OHads进行DFT 计算, 发现当In/Pd 原子比为3∶2 (即In3Pd2) 时, 颗粒表面的In 原子具有最低的OHads吸附能(-1.659 eV), 结果如图23 所示, 其中的数值表示OH 的化学吸附能. 因此, In 位点的存在促进了游离OH 的高效吸附, 从而加快了乙醛生成乙酸的反应速率(Pd 位点上EOR 的速率决定步骤). 此外, 组装成直接乙醇燃料电池后, In3Pd2/C 的开路电压(open circuit voltage, OCV) 比商业Pd/C 提高了0.25 V (从0.64 V 提高到0.89 V), 功率密度提高了约80% (从3.7 mW/cm2提高到6.7 mW/cm2), 说明其可作为替代Pt 或Pd 的催化剂. Chen 等[75]也提出了类似的观点, 向NiPd 合金中掺杂P, 制备得到PdNiP 合金纳米颗粒. 该研究认为, 由于Ni 和P 与Pd 之间的电子效应, 同时OH 在Ni 表面具有较强的吸附,使得EOR 的中间产物CH3CO 在Pd 表面很容易被邻近Ni 原子上吸附的OH 氧化, 得到乙酸产物, 提升了Pd 位点对EOR 的本征催化活性. 图23 Pd、In3Pd2 和In3Pd5 团簇表面OH- 物种的吸附模型Fig.23 Geometries of OH- adsorption complex on Pd, In3Pd2 and In3Pd5 clusters 合理的电催化剂设计需要耦合应变效应、配体效应和协同效应共同增强电催化活性. 这些效应在一定程度上都可以影响催化剂的d 带中心, 但是区分这些效应却并不容易. Sun等[76]合成了PdCuB 三元合金纳米催化剂. 该催化剂表现出了优异的电化学EOR 活性(质量活性为5.83 A/mgPd) 和良好的稳定性. 在KOH 和KOH/乙醇混合物中进行EOR 机理研究时发现, 引入的B 具有三重功能(见图24). 首先, B 修饰了Pd/Cu 的电子结构, 直接优化了乙氧基中间体在Pd 表面的吸附. 其次, B 可以促进OH 吸附, 从而在动力学上加速了乙氧基中间体的进一步氧化. 最后, B 的引入可以稳定Pd/PdCu 晶体, 提升了催化剂的稳定性. 图24 PdCuB 三元纳米合金催化乙醇氧化示意图Fig.24 Schematic diagram of ethanol oxidation catalyzed by PdCuB ternary nanoalloy 以上协同概念较少应用在ORR 强化过程中, 为了和本工作主题更为切合, 本工作描述了一种更为广义的协同, 试图拓宽协同效应的范围, 通过多种物理效应的协同, 实现电催化ORR的强化. 一些研究表明, Pt 族金属对反应物氧气分子及氧还原中间产物(例如H2O2、O/OH和等) 具有较强的键合能力, 其中Pd 金属具有比Pt 更高的键合能力[77-78], 使其ORR 催化性能相对较低. 因此, 通过降低Pd 金属的d 带中心, 减弱氧分子和中间产物与其活性位点的键合能力, 是提升Pd 催化ORR 性能的重要方式. 很多研究都发现生长在Au 内核颗粒表面的Pd 壳层在碱性介质中具有优良的ORR 催化性能, 其活性甚至可以超过Pt 纳米颗粒[79-83]. 这是因为相对Pd 金属(2.2), Au 元素具有较大的电负性(2.4), 能够造成Pd 原子的电子云向Au 内核转移. 这一电子耦合效应降低了ORR 过程的中间产物在Pd 表面的吸附能, 强化了其催化ORR 的性能. 但Au 颗粒也有不利的一面. Au 具有较大的晶格常数, 会对包覆在其表面的Pd 壳层施加拉伸应变效应. 该效应在Pd 壳层较薄时更加不能忽略, 能够使Pd 金属的d 带中心上移. 与电子耦合作用相反, 该效应强化ORR 中间产物在Pd 表面的吸附, 对ORR 产生不利影响. 为了利用核壳结构的优势并规避其不利影响, Zeng 等[84-85]发展了一个核壳结构与合金效应协同的策略, 即在保留电子耦合效应的同时, 一定程度上缓解晶格拉伸效应, 达到提升Pd催化ORR 性能的目的(见图25). 首先, 在Au 内核颗粒的表面形成一个超薄的过渡金属Ni壳层. 然后, 通过与Pd 前驱体进行伽伐尼置换反应(Galvanic replacement reaction, GRR),将Ni 壳层转化为超薄的NiPd 合金壳层. 这样, 在保留Au 内核与Pd 壳层电子耦合作用的同时, 具有较小晶格常数的Ni 原子掺杂对Pd 壳层产生了压缩效应, 一定程度上缓解了Au 内核施加的拉伸应变. 这两个效应协同作用, 使Pd 金属的d 带中心处在一个相对更加合适的位置,在ORR 中展现出了优良的电催化性能. 在这个协同策略中, 过渡金属Ni 可以更换为Fe、Co和Cu 等元素, 对Pd 金属的d 带中心都有类似的调节作用. 除了ORR 外, 也能在MOR 和EOR 中发挥强化作用. 图25 核壳结构协同合金效应提升Pd 催化ORR 性能示意图Fig.25 Schematic diagram showing the synergy of core-shell construction and alloy effect for enhancing Pd electrocatalysis in ORR 除了d 带中心, 其他描述符, 包括活性物质的表面浓度、配位数、键长、氢中间体的吸附能等, 也可用于描述特定电化学过程. 但这些描述符适用于单一的特定电催化反应, 缺乏对其他反应的普适性. 通过对催化剂内在性质和作用机制进行研究, 可以很好地衔接催化剂的设计与合成, 总结催化剂的共同性质, 得出可推广的、具有普遍性的规律和原则, 再将这些规律应用于指导催化剂的设计. 催化剂作为电催化的核心部分, 其设计与合成对于电化学的发展具有十分重要的意义, 因而受到广泛的关注. 本工作概述了燃料电池阴极发生的ORR 电极反应机理、涉及的催化剂性质调控和研究进展, 也从中看到了关于这类电极反应催化剂研究存在的困难与挑战: ①如何更有效缓解中间物种对Pd 基催化剂表面的毒害作用; ②如何调控d 轨道态密度中心, 使其处于合适的位置, 从而优化反应物种在Pd 催化剂表面的吸附; ③如何更好地耦合催化剂中各组分间的几何效应、电子效应、晶格应变效应和协同效应等, 发挥各种影响因素的优势, 调控Pd活性位点, 共同促进电催化反应的进行; ④如何提高活性组分Pd 的原子利用率, 降低催化剂的成本. 后续工作应考虑如何继续减少贵金属在催化剂中的用量, 例如制备单位点催化剂、发展单层贵金属壳的纳米催化剂等, 以降低催化剂的制备成本. 此外, 应着重筛选出几种催化剂进行透彻的表征与催化机制分析, 以深入挖掘影响催化性能的关键因素, 更好地修饰表面活性位点的性能, 也为其他种类高性能燃料电池催化剂的制备提供指导.

2.2 晶格应变效应




2.3 配体效应

2.4 协同效应


3 总结与展望
