Phelan-McDermid综合征42例病例系列报告
2023-12-15刘春雪邓晶鑫李慧萍张凯峰周秉睿胡纯纯
刘春雪 邓晶鑫 王 怡 李慧萍 张凯峰 董 萍 徐 琼 张 颖 周秉睿 胡纯纯 徐 秀
Phelan-McDermid 综合征(PMS,OMIM#606232)又称22q13.3缺失综合征,是一种罕见的发育障碍性疾病[1],通常由 22 号染色体长臂远端区域(包括SHANK3基因)的遗传物质丢失或SHANK3基因的致病性变异引起。PMS在中国或全球的患病率尚不清楚,至2022年,美国PMS基金会(https://www.pmsf.org/)登记人数已超过2 800人,SHANK3基因缺失或致病性变异估计频率为0.5%~2%[2]。国内关于 PMS的大样本报道很少[3, 4],大多为个案报告。本文收集复旦大学附属儿科医院(我院)儿童保健科近年来收治的PMS患儿的临床表型及遗传学特点,以提高临床医师对本病的认识和诊疗水平。
1 方法
1.1 PMS诊断 当患儿有新生儿肌张力减退、言语缺失或语言发育严重延迟、发育迟缓、轻微的畸形特征(包括长头、浓密的眉毛、面中部平坦、眼睛深陷、眼睑饱满、长睫毛、球状鼻、脸颊饱满、大耳)应怀疑PMS,并行基因检测,存在22q13缺失或SHANK3基因致病变异诊断为PMS。
1.2 病例纳入标准 2014年1月至2022年12月在我院儿童保健科通过基因检测明确22q13缺失或SHANK3基因致病变异诊断为PMS的连续病例。
1.3 PMS病例管理 本文作为2项国家自然科学基金项目(《SHANK3基因缺失/点突变与孤独症表型关系的研究及机制探讨》和《SHANK家族基因缺陷导致脑网络异常的多模态研究》)中PMS病例初诊的资料,在我院儿童保健科诊断的PMS均由专人负责临床表型系统采集和对PMS病例不定期线上和线下咨询和随访。
1.4 遗传学检查 22q13.3缺失检测时,先通过全基因组阵列比较基因组杂交检测来确认缺失片段的大小,再通过多重连接依赖性探针扩增(MLPA)技术确认SHANK3基因的缺失情况。由全外显子组测序或临床外显子组测序发现SHANK3基因点突变,并通过Sanger测序对患儿及其父母进行验证。根据美国医学遗传学与基因组学学会(ACMG)序列变异解读标准与指南对变异的致病性进行评估[5]。
1.5 PMS病例信息采集 从门诊病例中采集PMS患儿信息,①一般情况:母孕产史,患儿家族史、性别、我院初诊年龄、围生期情况、出生喂养史、生长发育史、既往史;②体格检查:身高、体重和头围,外部畸形特征(面容评估);③神经发育和行为评估:语言、发育评估(Griffiths/Gesell)、智商检测(韦氏评估)、孤独症谱系障碍(ASD)评估(DSM-5诊断标准+ADOS-2或ADI-R)、注意缺陷多动障碍(ADHD)评估(DSM-5诊断标准+多动症量表)等;④其他系统表现:过敏、反复感染、睡眠、消化功能、惊厥、肾脏、心脏、视听觉;⑤遗传学信息:PMS患儿及其父母SHANK3基因在内的22q13.3缺失信息。
2 结果
2.1 一般情况 42例PMS患儿进入本文分析,男24例(58%),女18例,我院平均初诊年龄为3.8(1.5~12.9)岁。1例早产儿(36+3周),41例为足月儿,其中2例为低出生体重儿(2 335 g和2 430 g)。
2.2 体格检查 42例PMS患儿中,3例(7%)身材高大(>+2SD),2例(5%)身材矮小(<-2SD),2例(5%)体重过重(>+2SD),3例(7%)低体重(<-2SD),2例(5%)头围过大(>+2SD),4例(10%)头围过小(<-2SD)。
2.3 畸形特征 42例PMS患儿中,与头长不相称大耳(图1A)但耳廓发育不良(耳朵很软)36例(86%),脸颊饱满34例(81%),鼻孔前倾朝上(图1B)28例(67%),眼睑饱满27例(64%),面中部平坦25例(60%),肉质手/脚22例(52%),球状鼻22例(52%),宽额头/额头突出21例(50%),内眦褶、赘皮20例(48%),牙齿发育不良或缺陷16例(38%),浓眉毛11例(26%),眼距宽8例(19%),长而浓密的睫毛、眉尾稀疏/缺如(图1C)和手/脚指甲发育不良各5例(12%),脚趾变形或交叠、长人中和体毛重各4例(10%),耳廓畸形(图1D)和骶骨酒窝各3例(7%),长头、垂眼眼型(图1E)和上睑下垂各2例(5%),眼睛深陷、高腭弓、小颌畸形、韧带松弛(图1F)各1例(2%)。
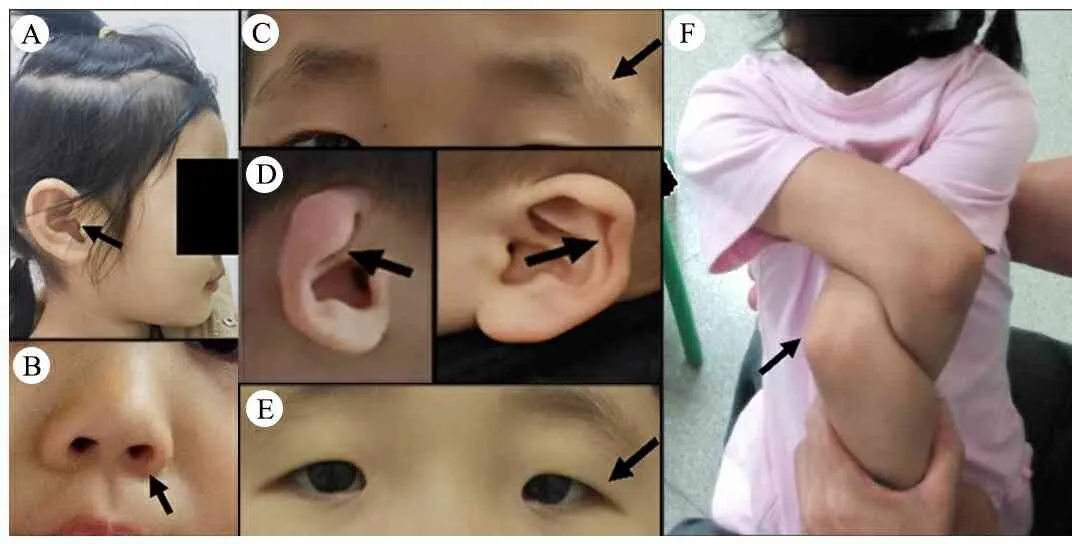
图1 PMS患儿畸形特征注 A:大耳(与头型不相称),B:鼻孔前倾朝上,C:眉尾稀疏/缺如,D:耳廓畸形,E:垂眼眼型,F:韧带松弛
2.4 神经发育和行为评估 42例均有语言发育延迟甚至语言缺失。其中无任何有意义单词27例(64%),会说单词(名词或动词)7例(17%),会说词组3例(7%),会说简单长句(缺乏复杂句)5例(12%);35例(83%)18月龄仍只会说无任何有意义单词,有意识叫人的平均年龄为21月龄。39例完成Griffiths量表评估(评估年龄1.5~8岁),与中国常模人群相比,运动能区92%
2.5 其他系统表现 42例PMS患儿中,咀嚼无力、喜欢吃软食和过敏(食物、药物)各18例(43%),反复感染(主要是呼吸道感染)16例(38%),睡眠障碍(如入睡困难、夜间易醒等)14例(33%),反复便秘12例(29%),较严重的偏食挑食11例(26%),血液系统疾病(包括贫血、特发性血小板减少性紫癜)9例(21%),惊厥史(5例热性惊厥,2例非热性惊厥)、肾脏发育异常和视觉问题(近视、散光、斜视)各7例(17%),蛛网膜囊肿6例(15%),反复腹泻史4例(10%),心脏发育异常3例(7%),听力异常2例(5%),周期性呕吐史和脉络膜囊肿各1例(2.5%)。
2.6 遗传学分析 根据ACMG序列变异解读标准与指南对变异进行分析,均为致病变异。均为SHANK3基因杂合变异,21例提示包含SHANK3基因在内的22q13.3缺失,缺失范围0.1~7.7 Mb,4例为SHANK3基因部分外显子缺失,17例为SHANK3基因杂合性突变,其中移码突变12例,无义突变5例[根据RefSeq中的SHANK3mRNA(NM_033517.1)和蛋白质(NP_277052.1)校正核苷酸和氨基酸位置]。39例为新发突变,3例为父母一方血样未获得(1例母亲和2例父亲阴性),42例PMS患儿遗传学信息见附件1(http://www.cjebp.net/CN/10.3969/j.issn.1673-5501.2023.05.010)。
3 讨论
PMS的特征包括全面发育迟缓/智力障碍、语言缺失或严重的语言发育障碍、肌张力减退、ASD、轻度的颜面部畸形特征、异常的行为特征等[6-8]。SHANK3基因(OMIM#606230)被认为是PMS神经学特征的关键候选基因,SHANK3是位于富含兴奋性突触的突触后致密区(PSD)的一种重要的突触支架蛋白,对突触的发生、成熟和功能的行使都至关重要[9-12]。SHANK3在形成突触后环境、整合突触后密度谷氨酸受体的蛋白质网络和维持突触结构方面发挥着核心作用[13]。在中枢神经系统和周围神经系统中,SHANK3蛋白与兴奋性突触的PSD中间支架蛋白相互作用,从而排列中间支架蛋白,影响突触发育和功能[14-18]。由DLG4(也称为PSD95)、DLGAP1(也称为SAPAP1或GKAP)、SHANK3和HOMER蛋白构建的PSD中的4级复合物连接代谢和离子型NMDA型谷氨酸受体复合物,共同调节突触的发育和功能连接[14-17]。各种Shank3突变动物模型表现出与人类患者症状相关的细胞和行为缺陷,如神经元形态改变(突触密度、长度降低)、神经元生理紊乱以及异常行为,包括社交互动、重复行为以及认知和运动功能障碍[10, 19-23]。本研究是目前国内最大样本的PMS系列研究报道。该疾病主要是染色体22q13的远端缺失引起,在大多数情况下包括SHANK3基因。80%的PMS患儿由22q末端缺失所致,PMS个体之间的缺失大小差异很大,SHANK3基因的个别外显子缺失从<13 kb到>9 Mb不等[8, 24, 25],缺失在男性和女性中发生的频率无明显差异[26]。在本研究中,缺失片段大小差异较大,从130 kb到7.7 Mb不等。
本研究42例PMS患儿中,最常见(>50%)的临床特征是全面发育迟缓/智力低下、语言发育障碍、ASD、注意力不集中、多动、大运动发育里程碑延迟、颜面部畸形(耳朵耳廓发育形成不良、脸颊饱满、鼻孔前倾朝上、眼睑饱满、面中部平坦、肉质手/肉质脚、球状鼻、宽额头或额头突出),还有一些较常见(20%~50%)的特征,包括咀嚼无力、过敏、技能退化、攻击性、反复感染、睡眠障碍、新生儿期肌张力低下、便秘、偏食挑食、血液系统问题。大部分临床特征与国外来自不同人群的报告基本一致[24-29](表1),但ASD、过敏、心脏发育异常、听力异常的发生率略高于国外报道,而反复腹泻、便秘、身材高大或矮小、头围过大或过小的发生率略低于国外报道。这些差异可能与样本量、种族、年龄尚小一些表型未出现以及临床资料不全等有关。国内Xu等[4]报道了29例,Hao等[3]报道了21例,Chen等[30]报道了10例(表1),由于样本量限制,难以得到一致性结论。

表1 42例PMS患儿体格发育情况、发育特征、异常行为及其他系统问题
值得关注的是,本文观察到的如下临床表型,体重过重(>+2SD)、低体重(<-2SD)、眉尾稀疏/缺如、体毛重、耳廓畸形、骶骨酒窝、垂眼眼型、上睑下垂、小颌畸形、韧带松弛、注意力不集中、大运动发育里程碑延迟(>18月龄)、偏食挑食、血液系统问题,在国外PMS病例报告最多的6篇文献(n=637)和国内PMS病例报告最多的3篇文献(n=60)中没有体现。
另外一些颜面部畸形特征在中西方人群中存在较大差异,如鼻孔前倾朝上的表型在国外仅一项研究报道[31];本文面中部平坦较国外报道高[2, 6, 25-27],这些可能与中西方人本身的面部外形差异有关,但全面发育迟缓(尤其是言语发育迟缓)/智力低下、ASD、肌张力低下是不同种族最常见的共同特征。
研究发现,PMS患者的技能退化最常发生在学龄期,主要影响运动和自理技能[32]。本研究中40%患儿经历过技能退化,主要涉及语言、社交和运动技能。照顾人应密切监测儿童在学龄期出现的技能退化,这样可以及早发现,并采取相应干预措施。PMS可以通过临床上技能退化来确定,这将促使转诊进行进一步基因检测从而更早期诊断PMS。干预措施可能有助于保持学龄期后期-青春期的技能或在退化后恢复技能,重要的是如何发展和应用这些干预措施[32]。
由于PMS临床表现缺乏特异性,易被误诊。目前尚未建立PMS的临床诊断标准,主要是依赖于遗传学诊断。本研究在综合国外大样本研究基础上,结合国内研究数据,建议在临床上,当新生儿期存在肌张力低下表现、明显的运动和语言发育里程碑的延迟,任何年龄段出现多个能区包括大运动、语言、手眼协调、个人-社会互动、表现等能区的发育严重落后,以及存在与头长不相称的大耳和与身材不相称的肉质手/脚的患者,应考虑PMS可能,并进一步完善相关遗传学检测来明确诊断。这将有利于早期发现、筛查可能受损的器官或脏器功能(如心脏、肾脏、神经、听力、视力等),早期干预训练,以提高患者的生存技能和认知水平。
本研究的不足之处在于,基于回顾性资料的分析和总结,缺乏个体的纵向随访数据,另外对于畸形特征缺乏具体数据的统计学分析。在未来研究中,我们将把PMS的主要临床特征量化,获得患者纵向发育和随访资料,并和其他综合征患者进行鉴别分析,制定PMS的临床诊断标准,从而减少诊断误差,提高诊断率。
