HIF-1α在胰腺导管腺癌发生发展中作用的研究进展
2023-12-13郭万慧彭雁飞孙力康
高 璠,郭万慧,韩 超,彭雁飞,周 昆,孙力康*
0 引言
胰腺导管腺癌(Pancreatic ductal adenocarcinoma,PDAC)在胰腺恶性肿瘤中最为多见,约占90%左右。PDAC通常在晚期被发现,与偶然发现的胰腺早期疾病相比,其确诊后的存活率很低[1]。美国癌症协会统计,预计2023年,因胰腺癌而死亡的人数高达50 550人[2]。近年来,多用异型增生来描述细胞增生并出现异型性,其与肿瘤形成相关且多用于上皮组织来源的病变[3]。原位癌是癌前病变,有恶性肿瘤的特点,但还没有突破基底膜向下浸润。目前,临床上采用上皮内瘤变(Intraepithelial neoplasia,IN)概括上皮的异型增生和原位癌。上皮内瘤变是癌症的高风险因素[4]。胰腺上皮内瘤变(Pancreatic intraepithelial neoplasia,PanIN)是最典型且常见的PDAC癌前病变,是胰腺导管腺癌的高风险因素[5]。随着对胰腺导管上皮增生性病变研究的深入,PanIN逐渐被临床、科研等领域接受,根据Hruban等[6]提出的胰腺上皮内瘤变分级系统和病理诊断标准,PanIN按严重程度从低到高分为PanIN-1(A,B)、PanIN-2和PanIN-3。其中,PanIN-3病变也称为原位癌,若进一步恶化,细胞突破基底膜后则称为胰腺导管腺癌。一旦确诊为癌症,5年生存率不足5%,因此,迫切需要寻找早期诊断指标。
胰腺导管腺癌的发生发展过程中伴随着典型的缺氧微环境,而缺氧诱导因子-1α (Hypoxia inducible factor-1,HIF-1α)作为缺氧环境的重要调节因子,参与调节PanIN各阶段相关基因,推动PanIN向PDAC进展。此外,在HIF-1α的调节下,肿瘤细胞还发生代谢重编程、血管新生、具有药物抗性以及迁移能力增强等改变。本文从PDAC腺癌前病变开始,探讨在PDAC的发生发展过程中,HIF-1α对PanIN及PDAC相关基因表达的影响。
1 HIF-1α简介
HIF-1是一种螺旋-环-螺旋转录因子,参与许多重要的缺氧稳态反应,通过控制转录反应来降低机体对氧气的利用率[7]。HIF-1是由2种亚基组成的异二聚体蛋白,这2种亚基分别是参与氧气调节的HIF-1α和控制结构性表达的HIF-1β。恶性肿瘤细胞的异常生长和增殖使得实体瘤普遍处于缺氧环境中,肿瘤细胞适应缺氧微环境主要依赖于HIF-1α介导的调节途径[8]。HIF-1α是癌症进展和靶向治疗的关键转录因子。在氧气充足的环境中,HIF-1α被泛素蛋白酶体途径(Ubiquitin proteasome pathway,UPP)降解失活。相反,在缺氧环境中,HIF-1α逃脱降解并进入细胞核,然后上调许多与癌症进展有关的基因[9]。过表达的HIF-1α和下游基因通过各种机制促进肿瘤发生发展,包括血管生成、细胞增殖和存活、代谢重编程、侵袭和转移、癌症干细胞维持、遗传不稳定性的诱导和治疗抗性。在肿瘤发生发展过程中,HIF-1α 还可以被与缺氧无关的信号通路激活[10]。
临床研究显示,在肿瘤发生前期HIF-1α出现积累[11-13]。与正常组织相比,肿瘤组织、IN组织中HIF-1α高表达;与低级别IN相比,高级别IN组织中HIF-1α表达水平增高;此外,在肿瘤附近的IN组织中检测到HIF-1α表达水平比独立IN组织中更高。结果表明,HIF-1α在IN发展过程中起到至关重要的作用,这可能与HIF-1α调节IN相关基因密切相关[13]。
2 HIF-1α与PanIN相关基因的相互作用
PanIN的发生率随着年龄的增长而增加,与老年人群中胰腺癌发病率的增加相对应[14]。基于对人体组织样本的回顾性分析,推测胰腺外分泌细胞中积累了一系列基因组突变,导致胰腺上皮内瘤变,然后发展为侵袭性PDAC[15]。但并不是所有低级别病变都向高级别病变发展,在发展过程中,存在部分高级别上皮内瘤变向低级别转归的现象。根据基因改变出现的时间节点不同,研究者将其分为早期改变和晚期改变基因。早期改变有原癌基因KRAS突变、粘蛋白MUC5AC过表达、抑癌基因P16失活及粘蛋白MUC1表达变化等;晚期改变有抑癌基因TP53突变等[16-18]。见图1。HIF-1α对以上基因的调节作用见图2。
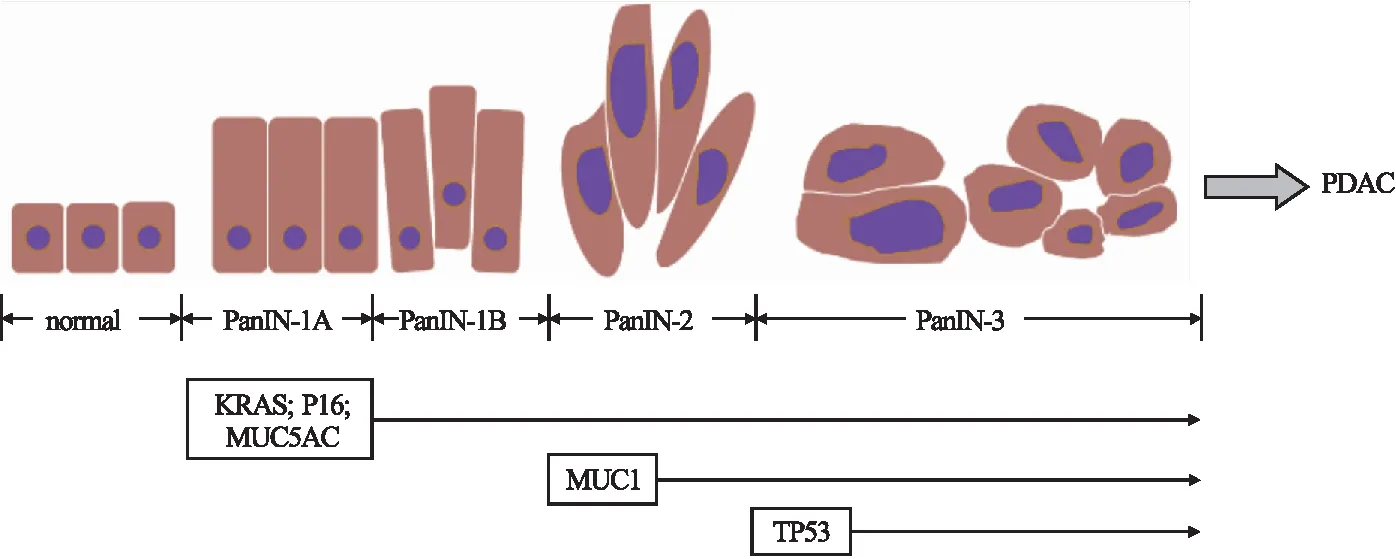
图1 胰腺上皮内瘤变的发展阶段及突变基因
2.1 HIF-1α与KRAS之间的正反馈调节KRAS(Kirsten rat sarcoma virus)是为制造K-Ras蛋白质提供指令的基因,是RAS/MAPK途径的一部分。KRAS原癌基因的突变是PanIN逐步进展过程中的重要因素[19]。研究表明,随着PanIN级别升高,KRAS突变频率增加,人类PDAC中几乎普遍存在KRAS突变(>95%)[20]。临床研究显示,与KRASG12R突变相比,KRASG12D突变的PDAC患者总体生存率降低[21]。研究者采用p48-Cre诱导KRAS突变,获得从正常胰腺到胰腺导管腺癌发展各阶段的基因工程小鼠模型,在小鼠2月龄时,其胰腺组织结构正常,在小部分区域出现上皮内瘤变。在野生型样本中,低氧探针(pO2水平≤1% 的指标)和HIF-1α蛋白几乎检测不到,而在2个月大的KRASG12D小鼠胰腺组织中观察到低氧探针阳性细胞和HIF-1α表达显著增加,表明胰腺上皮内瘤变过程也处于低氧环境,KRASG12D突变促进HIF-1α积累[22]。此外,HIF-1α或缺氧条件可以上调KRAS活性及其下游信号的激活[23]。因此,在KRAS激活和HIF-1α之间形成了正反馈调节。
上述结果提示,HIF-1α与KRAS2个信号共同作用,促进PanIN向更高级别进展。
2.2 HIF-1α上调粘蛋白MUC5AC的表达 粘蛋白(Mucoprotein)是高分子糖蛋白,在正常的呼吸道和消化道形成一层屏障,保护下层上皮组织免受炎症、感染、酸(胃肠道内)和其他生理损伤[24]。MUC5AC是一种重度糖基化的凝胶形成分泌粘蛋白,在多种恶性肿瘤中具有可靠的预后价值[25]。大量研究表明,MUC5AC在正常胰腺导管内不表达,但在PanIN-1A阶段表达,且在PDAC和癌前病变中均以高水平表达[26-28]。在缺氧条件下,HIF-1α水平的升高能够显著促进MUC5AC蛋白表达。在常氧条件下,使用HIF-1α抑制剂或HIF-1α siRNA下调HIF-1α的水平后,MUC5AC的分泌与蛋白表达也随之下调。表明无论在RNA水平还是蛋白水平上,HIF-1α均能干预MUC5AC[29-30]。而且,缺氧条件下MUC5AC启动子活性增加,荧光素酶测定表明,MUC5AC启动子中的缺氧反应元件(Hypoxia-response element,HRE)在-120~+54区域。启动子序列分析表明,-65处的HRE位点在MUC5AC的缺氧激活中起重要作用。使用定点诱变使HRE部位失活导致缺氧诱导完全丧失,进一步证实了MUC5AC启动子中的缺氧反应元件HRE部位在缺氧环境中的关键作用[30]。结果显示,缺氧或可通过上调HIF-1α表达,进而上调粘蛋白MUC5AC的表达,促进胰腺上皮内瘤变进展。
2.3 肿瘤抑癌基因P16抑制HIF-1α的表达 肿瘤抑癌基因P16亦称为多肿瘤抑制因子(Multiple tumor suppressor,MTS),是细胞周期中的基本基因,直接参与细胞周期的调控。肿瘤抑癌基因P16失活发生在PanIN-1B阶段[27]。P16位于9p21,是多种恶性肿瘤杂合性丢失(Loss of heterozygosity,LOH)的位点,因此P16与多种肿瘤有关[31]。P16与KRAS、TP53、SMAD4(该基因编码信号转导蛋白Smad家族的1个成员,Smad蛋白被跨膜丝氨酸-苏氨酸受体激酶磷酸化和激活,以响应转化生长因子TGF-β信号)的失活是人类PDAC中最常见的基因突变。这些突变基因是胰腺癌中涉及的“四大”基因[32]。研究者用小干扰RNA干扰P16表达后,AKT/mTOR通路随之被激活;同时,HIF-1α表达上调,进而上调血管内皮生长因子(Vascular endothelial growth factor,VEGF)。结果表明,HIF-1α是P16基因的下游靶点,当抑癌基因P16失活后,HIF-1α逐渐积累并调控下游促肿瘤因子,如VEGFA等,从而促进胰腺上皮内瘤变的发展[33]。
2.4 粘蛋白MUC1上调HIF-1α的表达 人MUC1粘蛋白是一种膜结合糖蛋白,是正常腺细胞导管细胞表面的主要成分。MUC1在癌细胞中过度表达并异常糖基化[34]。Maitra等[35]研究表明,MUC1在正常小叶内和小叶间胰腺导管中100%表达,在PanIN-2和PanIN-3中的表达分别为43%和85%,但在PanIN-1A和PanIN-1B中只存在5%、6%的表达。因此,在胰腺癌的进展过程中,MUC1从低表达到过表达,其表达由正常极化上皮细胞的顶端变为广泛分布于非极化肿瘤细胞的细胞膜表面。MUC1通过调节HIF-1α的表达、稳定性和活性,在胰腺癌细胞中作为低氧反应的调节剂。Chaika等[36]研究显示,MUC1在物理上占据参与糖酵解的多个基因的启动子并调节关键代谢酶活性。在此基础上,MUC1表达促进胰腺癌细胞的葡萄糖摄取。此外,HIF-1α是吉西他滨耐药的胰腺癌细胞(Gem-R细胞)葡萄糖代谢和嘧啶生物合成增强的代谢主调节因子,抑制HIF-1α的表达可提高细胞对吉西他滨的敏感性。Shukla等[37]研究显示,MUC1增强HIF-1α表达水平并降低Gem-R细胞对吉西他滨的敏感性。上述研究表明,MUC1不仅可以上调HIF-1α来促进胰腺肿瘤细胞代谢重编程能力,以适应胰腺癌的缺氧环境;还可以通过上调HIF-1α增强胰腺癌细胞的耐药性。在MUC1过表达进而促进肿瘤发生发展的过程中,HIF-1α起重要的纽带作用。
2.5 抑癌基因 TP53下调HIF-1α的表达 TP53抑癌基因位于染色体臂17p上,是人类实体肿瘤中最常失活的抑癌基因,在55%~75%的侵袭性胰腺癌中失去抑癌活性[38]。TP53突变的发生率随着癌前病变程度的增加而增加,在低级别PanIN中发生率非常低,但在高级别PanIN中发生率显著升高[39]。TP53肿瘤抑制因子可以被各种细胞应激反应激活,包括癌基因表达、DNA损伤、缺氧、代谢功能障碍等,然后以适当的反应来抑制癌症的发生[40]。在常氧环境中,HIF-1α在脯氨酸残基处被如von Hippel-Lindau 抑癌基因蛋白(VHL)和 MDM2 (Mouse Double Minute 2 homolog)等蛋白泛素化,最终HIF-1α被蛋白酶体降解[41]。突变后的TP53 (Mutant p53,mutp53)可以在缺氧条件下破坏HIF-1α与泛素蛋白连接酶MDM2的结合。研究显示,癌细胞中HIF-1α及其靶基因的高表达可能是mutp53解离HIF1α-MDM2的直接后果[42]。上述结果表明,HIF-1α作为P53的靶基因,参与调控胰腺腺泡细胞导管样化生及PanIN进程,促进PanIN-3向PDAC恶性转化。
3 HIF-1α与PDAC相关基因的相互作用
胰腺癌是全球癌症相关死亡的主要原因之一,其特征是高度缺氧的肿瘤微环境。HIF-1α是细胞对氧浓度变化反应的主要调节因子,帮助肿瘤细胞在缺氧肿瘤微环境中适应缺氧。HIF-1α在胰腺癌的癌变和进展中起核心作用,其与PDAC相关基因的相互作用见表1。

表1 HIF-1α与PDAC相关基因的相互作用
3.1 HIF-1α与赖氨酸特异性去甲基化酶(Lysine-specific demethylase 1,LSD1)之间的正反馈调节 LSD1也称为KDM1A、BHC110、AOF2,是首个被发现的人类组蛋白去甲基化酶[43],在许多癌症中均高表达,包括胰腺癌、乳腺癌、前列腺癌、食管癌、膀胱癌、肺癌、神经母细胞瘤和急性髓系白血病[44]。在胰腺癌细胞中,LSD1具有癌基因功能,参与调控促进细胞增殖相关基因,从而促进体外细胞迁移和侵袭[45-46]。对48例人体胰腺癌组织样本进行免疫组织化学染色分析的研究显示,与正常组织相比,胰腺癌组织中,LSD1表达增加,且其表达增加与胰腺癌患者的不良预后相关。此外,LSD1敲低的胰腺细胞葡萄糖摄取和乳酸生成也显著降低。HIF-1α稳定表达会上调LSD1的mRNA和蛋白水平,LSD1表达降低也会导致 HIF-1α靶向限速糖酵解酶在转录水平下调。总之,在缺氧条件下,LSD1和HIF-1α相互调节并形成正反馈回路,以增强细胞糖酵解能力,从而促进胰腺癌细胞不受控制的增殖。
3.2 HIF-1α上调血VEGF VEGF蛋白家族包括VEGF-A、VEGF-B、VEGF-C、VEGF-D、VEGF-E和胎盘生长因子(Placental growth factor,PlGF),其中,鉴于VEGF-A在调节血管生成及疾病中起主导作用,研究者通常将其称为VEGF[47]。与正常胰腺和慢性胰腺炎相比,VEGF在胰腺癌中过度表达,在93%的胰腺导管腺癌中VEGF 蛋白呈阳性。VEGF在胰腺癌组织中的阳性表达率为77%,在邻近的正常胰腺组织中只有 15%[48-49]。VEGF是HIF-1α的下游靶标,缺氧状态下,VEGF的表达主要受HIF-1α的转录水平调节[50]。HIF-1α上调后,VEGF分泌升高对于肿瘤的发生发展起至关重要的促进作用。HIF-1α表达与VEGF表达呈正相关。在胰腺癌细胞中,采用转染技术敲低BXPC-3细胞中HIF-1α后,VEGF表达显著下调[51],影响肿瘤微环境,进一步抑制肿瘤血管新生,从而降低胰腺癌细胞的侵袭、迁移能力。
3.3 HIF-1α上调内质网保留因子1 (Retention in endoplasmic reticulum-1,RER1) RER1是多种蛋白质在内质网中的重要保留因子,其与许多蛋白质相互作用,并将它们招募到COPⅠ囊泡,从而转移至内质网[52]。RER1不仅与胰腺肿瘤细胞干性相关,还促进胰腺癌细胞转移。与正常胰腺导管上皮细胞hTERT-HPNE相比,胰腺癌细胞中RER1的表达更高。研究显示,与未发生淋巴结转移的胰腺癌患者相比,RER1的表达显著增高,RER1高表达可能与胰腺癌患者预后不良有关,RER1 表达较高的患者的复发率更高,生存率更低[53]。缺氧环境下,HIF-1α无法通过泛素化降解,从而在体内堆积,随后RER1的表达也被上调。研究显示,肿瘤上皮间质转化相关基因也受RER1调节,与其呈正相关[53]。总之,缺氧诱导HIF-1α表达促进RER1表达,从而调节胰腺癌上皮间质转化相关基因,促进肿瘤细胞转移的重要环节——上皮间质转化[54]。
3.4 HIF-1α下调白细胞介素IL-37 IL-37已被确定为先天免疫和适应性免疫的抑制剂[55]。研究表明,IL-37可以依赖于 caspase-1 的方式易位到细胞核中,从而减少细胞因子的产生并影响先天性和适应性免疫反应[56]。在炎症环境中,IL-37通过下调焦亡相关蛋白,抑制急性胰腺炎小鼠腺泡细胞死亡[57]。在胰腺癌患者样本中,与邻近的正常胰腺组织相比,PDAC组织中的IL-37表达显著降低。PDAC中IL-37表达降低与PDAC组织学分级、肿瘤大小、淋巴结转移和血管浸润增加相关。低表达IL-37患者的无复发生存期和总生存期显著缩短[58]。此外,在胰腺癌细胞耐药性的研究中,IL-37 表达与吉西他滨疗效呈正相关,而在吉西他滨耐药性强的细胞系中,HIF-1α表达上调,研究人员通过IHC染色探究人 PDAC 样本中HIF-1α和IL-37的表达情况,结果显示,在 PDAC 的连续切片中,HIF-1α和IL-37表达呈负相关[58],表明HIF-1α可能通过与IL-37启动子中的缺氧反应元件结合来减弱IL-37转录,降低IL-37表达水平,从而促进PDAC对吉西他滨的耐药性及迁移能力。
4 小结
HIF-1α调控多种血管新生的基因,在生理状态下发挥促进血管生成的作用,HIF-1α激动剂在缺血性疾病方面有积极的治疗作用[59]。但在肿瘤中,HIF-1α作为缺氧环境中的关键调节因子,对PDAC的影响贯穿了整个发生发展过程。通过不同的方式抑制HIF-1α的活性,对于恶性肿瘤的治疗具有重要意义[60]。对PDAC癌前病变相关基因差异表达的研究显示,从正常组织到胰腺上皮内瘤变组织再到胰腺导管腺癌组织中,HIF-1α的表达水平越来越高[11]。此外,在胰腺上皮内瘤变的发展过程中,不断有肿瘤调控相关基因发生差异表达,其中具有代表性的有早期改变基因:原癌基因KRAS、粘蛋白MUC5AC、抑癌基因P16及粘蛋白MUC1等;晚期改变基因:抑癌基因TP53。这些发生改变的基因与HIF-1α互相作用,提示HIF-1α可能通过调控PanIN各阶段相关基因,进而调控PanIN进程。此外,HIF-1α还从多个角度参与促进PDAC的发展,其中包括帮助肿瘤细胞代谢重编程、血管生成、促进胰腺癌上皮间质转化以及增强肿瘤细胞对吉西他滨的耐药性等。
胰腺位置隐匿,检测手段有限,缺乏有效的早期诊断,使PDAC总体5年生存率极低。目前,首选的治疗方案仍是手术切除或以吉西他滨为代表的药物进行化疗,但效果仍然有限,迫切需要开发早期诊断和治疗工具[61]。与胰腺导管腺癌相似,宫颈癌也是从上皮内瘤变阶段发展而来,但目前对于宫颈癌已有大量研究[62-64],对宫颈上皮内瘤变的转归比例及发展时间都有较明确的结论。宫颈上皮内瘤变(Cervical intraepithelial neoplasia,CIN)分为3个阶段,约有60%的CIN-1自然消退,55%的CIN-2自然消退,28%的CIN-3自然消退,仅有少部分发生进展[65];且从CIN-3阶段发展到具有侵袭性的宫颈癌需要10~20年。关于PanIN的转归比例、发展时间以及基因调控途径尚不明确,但可以确定在PanIN发展过程中存在复杂的基因调控网络,促进PanIN向高级别进展,进而恶化成PDAC[28,66]。HIF-1α在PDAC发生发展过程中起重要作用,其与PanIN相关基因的互相调控关系值得深入研究。
