MoS2 中S 原子空位形成的非绝热动力学研究*
2023-12-01王月马杰
王月 马杰
(北京理工大学物理学院,先进光电量子结构设计与测量教育部重点实验室,北京 100081)
缺陷是半导体领域中最核心的问题.采用含时密度泛函方法,模拟了S 原子脱离MoS2 晶格形成空位缺陷过程中的电子动力学行为,发现该过程中存在显著的非绝热效应.非绝热效应导致S 原子需要消耗更多能量以脱离晶格形成空位缺陷.随着S 原子的初始动能增大,其脱离晶格形成空位的能量势垒也持续增大,并且在初始动能达到22 eV 附近时发生了阶跃式的增长.这是由朗道-齐纳电子跃迁和能级间库仑作用共同导致的.非绝热效应还改变了脱离晶格的S 原子上电荷的轨道分布,以及晶格中缺陷附近的电荷分布.此外,还发现该过程中自旋轨道耦合十分重要,必须被考虑.本文阐明了MoS2 中S 原子空位的形成机制,尤其是电子非绝热动力学的重要作用,为进一步研究缺陷对材料物理性质的调控提供了理论基础.
1 引言
MoS2作为二维过渡金属硫属化物[1]中半导体材料的代表,因其优异的理化性能和广阔的应用前景受到科研人员的持续关注.MoS2具有高载流子浓度、强自旋轨道耦合(spin-orbit coupling,SOC)等特点[2,3],在多种前沿基础研究领域受到重视,例如光电子学、谷电子学和自旋电子学等[3—6].在应用方面,MoS2也可以作为核心材料用于多种应用场景,例如场效应晶体管[7]、传感器[8]、柔性产品[3]和析氢反应[9]等.实际应用中MoS2材料往往是含有缺陷的[10].缺陷能改变MoS2的电子结构,例如在带隙中引入缺陷态[11,12]、改变局部载流子类型[6,11,13]以及降低肖特基势垒高度[14,15],能影响材料的光学响应[16]、增强MoS2的析氢催化性能[17]等.缺陷的出现虽然一定程度上改变了材料的固有属性,但是也为人为改造材料理化性质,使之朝着更适合人们需求的方向发展提供了可能性[10,18,19].
实验上发现MoS2中最主要的缺陷类型为S 原子空位缺陷[11].然而,Komsa 等[20]指出,在透射电子显微镜(transmission electron microscopy,TEM)实验中观察到的S 原子空位缺陷可能不是MoS2中固有的,而是在实验过程中人为造成的.这是因为TEM 在扫描材料结构的过程中,其高能电子束会对MoS2晶格造成损伤,使S 原子脱离晶格.这种由电子束辐射导致的S 原子空缺有3 种形成机制: 电子与原子之间弹性碰撞导致的原子散射,又称为“knock-on”损伤[20,21];非弹性电子散射诱导的辐射分解和电离[22—24];化学蚀刻[25,26].当电子束能量较高时,S 原子空位缺陷主要由弹性碰撞引起[27].碰撞发生后,S 原子从电子束获得一定的动能.当这一能量足够大时,原子将脱离晶格、不再复合,即在MoS2晶格中留下一个空位缺陷.分子动力学研究表明,S 原子脱离晶格所需的最小初始动能(“knock-on”阈值能量)为6.9 eV,与系统的空位形成能基本相等[20].但是,分子动力学是基于绝热近似的,在模拟过程中电子始终处于基态,因此体系的势能只与原子间相对位置有关而与初始动能无关.在实际过程中,由于S 原子有很高的初始动能,S 原子脱离晶格形成空位必然是一个非绝热过程,即电子会被激发到高能量的激发态.非绝热效应也将影响包括S 空位形成的阈值能量在内的一系列物理性质,因此采用非绝热的模拟方法研究该过程十分必要.
本文基于含时密度泛函理论(time-dependent density functional theory,TDDFT)[28],对单层MoS2体系中S 原子空位缺陷的形成过程进行了一系列非绝热的模拟与分析研究.通过改变S 原子脱离晶格的初始动能,研究了该变量对系统能量转化、本征能级变化、电子占据、电荷分布等物理性质的影响,发现在该过程中存在的朗道-齐纳(Landau-Zener,LZ)电子跃迁现象是造成上述影响的根本原因.通过比较非绝热模拟和绝热模拟结果之间的差异,揭示了空位形成过程中的微观机制,尤其是非绝热效应对电子动力学和缺陷形成的影响.
2 计算方法
2.1 计算模型
计算都采用第一性原理计算包PWmat[29,30]实现.为研究非绝热效应,采用PWmat 的实时含时密度泛函理论(real-time TDDFT,RT-TDDFT)计算模块进行模拟.在所有模拟中,采用局域密度近似(local density approximation,LDA)[31]交换关联泛函和ONCVPSP (optimized norm-conservation Vanderbilt pseudopotential)[32]模守恒赝势.若后文无特殊说明,模拟中均考虑SOC.在RTTDDFT 计算中时间步长取为0.1 fs,波函数截断能量为36 Ryd (1 Ryd=13.6057 eV),k点抽样只选取单Γ 点.图1(a)为单层MoS2的结构,为研究S 原子空位的形成,搭建了5 × 5 的超胞.
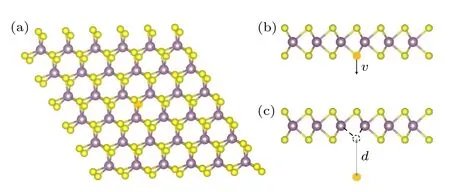
图1 (a) 5 × 5 的完整MoS2 超胞;(b) 发生弹性碰撞后,S 原子获得一定的初始速度v 脱离晶格,其速度方向垂直于晶格;(c) S 原子脱离晶格后运动到距离晶格为d 的位置.图中黄色球与紫色球分别代表S 原子与Mo 原子,高亮的原子为脱离晶格的S 原子Fig.1.(a) The 5 × 5 supercell of MoS2;(b) after the elastic collision with electrons,the S atom gets an initial velocity v,whose direction is perpendicular to the lattice;(c) the moving distance d of the S atom after sputtering from the lattice.Yellow and purple balls represent the sulfur and molybdenum atoms,respectively.The highlighted ball represents the sputtered S atom.
2.2 初始动能的选取
当完整的MoS2晶格受到高能电子束的轰击后,表面的S 原子将获得一定的动能,从而有可能脱离晶格并形成空位.内部的Mo 原子由于受到两侧S 原子层的阻挡,无法挣脱晶格[20].因此,只讨论S 原子空位形成的过程,即在模拟过程中只赋予一个S 原子以较大的初始动能,如图1(b)所示.
高能电子束与S 原子的碰撞过程符合相对论两体碰撞模型.本文只考虑S 原子初始速度垂直于MoS2平面的情况,如图1(b)所示.此时原子获得的最大动能符合[21]:
其中,Ek为碰撞后S 原子的动能,也是S 原子在脱离晶格模拟过程中的初始动能.它由入射电子动能Ee和碰撞前S 原子在晶格中的振动速度v0共同决定.me与ms分别为电子和S 原子的质量,c为光速.
根据密度泛函计算,MoS2系统中S 原子脱离晶格形成空位所需的最小动能约为6.9 eV,相应的高能电子束能量为80—90 keV[20,27].为了确保模拟中S 原子能完全脱离晶格而不发生复合,将S原子的最小初始动能取为7.6 eV.通常,TEM 中的电子束电压上限可达300 kV,且MoS2材料在该辐射能量下仍不会遭到严重损坏[20,33],此时S 原子获得的初始动能接近30 eV.因此,在7.6—30.4 eV内选取了不同的S 原子初始动能,进行了一系列TDDFT 模拟,以探究S 原子脱离晶格形成空位过程中的电子动力学行为,及其对缺陷形成的影响机制.
3 结果与讨论
3.1 S 原子脱离晶格的能量势垒
首先在绝热近似下模拟了MoS2中S 原子脱离晶格的过程,其能量势垒为6.9 eV,且不依赖于初始动能,与文献[20]中的结果一致.接着,采用TDDFT 方法,非绝热地模拟了MoS2中S 原子以不同初始动能脱离晶格束缚的过程,系统(包括MoS2晶格与脱离晶格的S 原子)势能Ep随S 原子运动距离d的变化关系如图2(a)所示.在非绝热模拟中,S 原子脱离晶格的势垒明显依赖于初始动能.
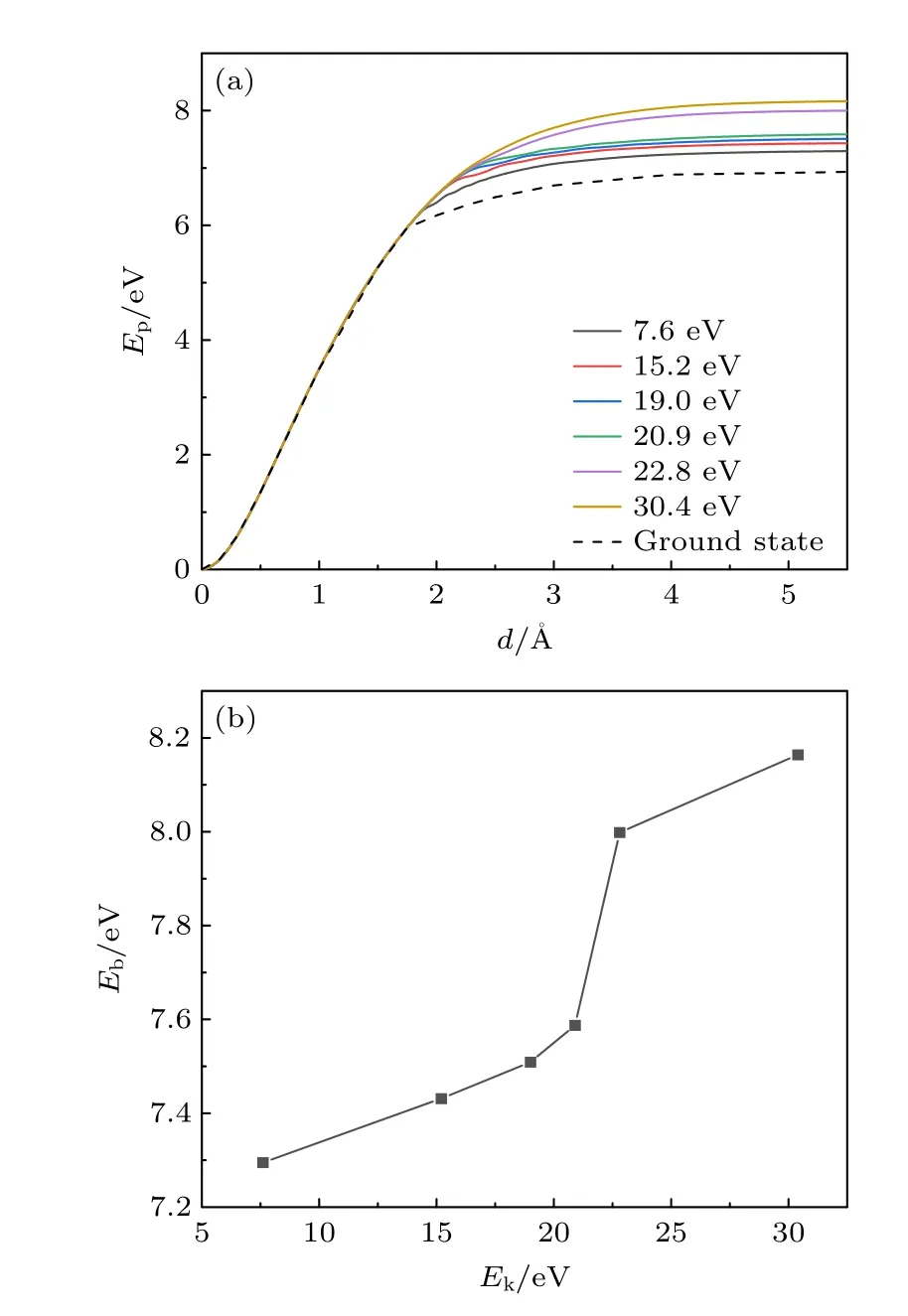
图2 (a) 当S 原子以不同初始动能脱离晶格时,系统势能Ep随S 原子运动距离d 的变化关系,其中初始时刻完整MoS2晶格的情况为势能零点;(b) S 原子脱离晶格所需越过的能量势垒与初始动能的关系Fig.2.(a) For the sulfur atom with various initial kinetic energies,the energy Ep as a function of the atom distance d,where the energy of the perfect MoS2 lattice is set to zero;(b) energy barriers for the S sputtering process as a function of the initial kinetic energy.
由图2(a)可知,随着S 原子逐渐远离MoS2,不论是绝热模拟还是非绝热模拟,系统的势能均先显著增长后趋于稳定.这是由于原子脱离晶格时,其动能不断转化为势能.当原子距离晶格较近(d<2 Å)时,S 原子与MoS2两者间的相互作用较强,Ep持续增长,能量变化明显;当原子距晶格较远(d> 2 Å)时,相互作用逐渐减弱,Ep变化放缓,并最终达到稳定.此外,可以看到,当d< 2 Å时,绝热模拟与非绝热模拟给出的势能曲线重合,这表明此时不存在明显的非绝热效应,Ep的大小与S 原子脱离晶格的初始动能无关,只与原子的运动距离有关.而当d> 2 Å时,非绝热模拟给出的势能曲线与绝热模拟的结果不再重合,且不同初始动能所对应的曲线也不再相同,即存在明显的非绝热效应.与绝热模拟相比,非绝热模拟下势能的增长幅度明显变大.由此可知,非绝热效应推高了系统的能量.S 原子的初始动能越大,相应非绝热模拟所得的系统势能也越大,即非绝热效应越明显.
当S 原子与晶格的距离d> 4 Å时,势能曲线基本不再变化,此时S 空位缺陷已经形成.缺陷形成后与缺陷形成前(d=0 Å),系统势能的差值即为S 原子脱离晶格的能量势垒Eb.Eb与S 原子初始动能Ek的关系如图2(b)所示,Eb随Ek增加而增加.S 原子初始动能在7.6—30.4 eV 之间时,其相应的能量势垒在7.3—8.2 eV,变化幅度约为1 eV.所有非绝热模拟给出的能量势垒均高于绝热模拟给出的6.9 eV.这表明当考虑非绝热效应后,S 原子将消耗更多的能量以脱离MoS2晶格的束缚,且初始动能越大的S 原子在脱离晶格的过程中消耗的能量越多.此外,还可以注意到,当S 原子初始动能不超过20.9 eV 时,随着初始动能的增长,Eb增幅基本保持线性;而当初始动能从20.9 eV增至22.8 eV 时,Eb曲线出现阶跃式增长;随后,其增速再次放缓.
3.2 非绝热跃迁
首先忽略SOC,在非绝热模拟中,电子能级随S 原子距离d的变化关系如图3(a)所示,红色气泡表示该能级被电子占据,气泡大小表示电子的占据数.随着脱离晶格的S 原子的运动距离d不断增加,初始时价带里的两条简并的能级能量上升(这两条能级主要由脱离晶格的S 原子的px和py轨道构成),而导带里的一条能级能量下降(这条能级主要由脱离晶格的S 原子的pz轨道构成).在d=1.9 Å时,以上3 条能级的能量发生交叉.我们注意到能级交叉时并未打开能隙,这表明这些能级相互之间不耦合.能级交叉后,两条简并的px和py能级的能量继续上升,而pz能级的能量继续下降.由于能级间不耦合,电子不会在能级间跃迁,因而px和py两条能级一直是满占据的,而pz能级一直是空的.

图3 系统能级随S 原子运动距离d 的变化情况.能级上的红色气泡表示该能级被电子占据,气泡大小表示电子占据数.插图为箭头所指能级的电荷分布 (a) 不考虑SOC;(b) 考虑SOC 且脱离晶格的S 原子的初始动能较低(Ek=15.2 eV);(c) 考虑SOC 且脱离晶格的S 原子的初始动能较高(Ek=30.4 eV)Fig.3.Energy levels of the system as a function of the atom distance d. Red bubble on an energy level indicates that the energy level is occupied by electrons,and the size of the bubble indicates the number of electrons occupied.The insets show the charge densities of the corresponding states: (a) Without SOC;(b) with SOC and a low initial kinetic energy of the S atom (Ek=15.2 eV);(c) with SOC and a high initial kinetic energy of the S atom (Ek=30.4 eV).
考虑SOC 后,电子能级随S 原子距离d的变化如图3(b)和图3(c)所示.与无SOC 的情况类似,随着d的不断增加,初始时价带里的两条双重简并能级的能量上升,而导带里的一条双重简并能级的能量下降.但与无SOC 情况不同的是,当这些能级发生交叉时打开了带隙,这表明能级之间存在耦合.在能级交叉前,能量低的两条能级由脱离晶格的S 原子的px和py轨道构成,能量高的能级则由脱离晶格的S 原子的pz轨道构成;能级交叉后,由于能级间存在耦合,其轨道成分也出现混合,能量最低的能级的主要成分是脱离晶格的S 原子的pz轨道,其他能量高的能级的主要成分是脱离晶格的S 原子的px和py轨道,即交叉前后能级顺序发生反转,这也与无SOC 的情况相同.此外,发现px和py能级中只有一条和pz能级耦合,另一条pxy能级不参与耦合.不参与耦合的能级一直是满占据,而电子可以在相互耦合的能级间发生跃迁.
电子的这种非绝热跃迁可以用LZ 跃迁模型描述[34,35].对于两个随绝热参数x变化的本征能级,含时薛定谔方程可以表示为
其中Φ1,2为这两个本征态,Ω为两态之间的耦合系数;E1,2为无耦合(Ω=0)时两本征态所对应的本征能量,并且其值在xc处相等.当考虑耦合后,两本征能级在xc处不再相等.反映在能级演化图像上,原本发生交叉的本征能级在考虑耦合后打开了一定宽度的能隙.
如果电子在远离xc处时处于某一本征能级上,当逐渐靠近并越过xc点时,电子有机会从该能级跃迁至另一能级,其跃迁几率PLZ符合
其中,β为两本征能级在xc处斜率的差,即
为了更清晰直观地阐述S 空位形成过程中电子的跃迁规律,给出了不同原子运动速度条件下,相关能级上的电子数目变化情况,如图4 所示.低速时(图4(a),S 原子的初始动能为15.2 eV),能量高的能级在交叉前(此时能级的主要成分为脱离晶格的S 原子的pz轨道)电子占据数为0,当S 原子运动距离d=1.8 Å附近时开始出现电子占据,当d> 2.4 Å后(此时能级的主要成分为脱离晶格的S 原子的pxy轨道)大约有0.28 个电子占据.相应地,能量低的能级在能级交叉前(此时能级的主要成分为脱离晶格的S 原子的pxy轨道)是满占据的,在交叉后(此时能级的主要成分为脱离晶格S 原子的pz轨道)电子占据数约为1.7.比较能级交叉前后pz轨道和pxy轨道上的电子占据数可知,存在约1.7 个电子从pxy轨道跃迁至pz轨道.在绝热近似下,能量高的能级上应无电子占据而能量低的能级上电子应满占据,因此在S 原子脱离晶格形成空位缺陷的过程中存在显著的非绝热效应.此外,电子的占据情况与不考虑SOC 时截然不同.这也表明,尽管在整个过程中系统没有产生磁矩、时间反演对称性没有被打破、所有能级都是二重简并的,但SOC 仍然起了很重要的作用,因而必须被考虑.

图4 (a) 当脱离晶格的S 原子初始动能较低时(Ek=15.2 eV),相关能级上电子占据数的变化;(b) 当脱离晶格的S 原子初始动能较高时(Ek=30.4 eV),相关能级上电子占据数的变化;(c) 原子运动距离d=3 Å时,高能量能级上的电子占据数随S 原子初始动能 Ek 的变化Fig.4.(a) When the initial kinetic energy of the sputtered S atom is low (Ek=15.2 eV),the electron occupations on the relevant states as functions of the atom distance;(b) when the initial kinetic energy of the sputtered S atom is high (Ek=30.4 eV),the electron occupations on the relevant states as functions of the atom distance;(c) electron occupation on the high-energy state as a function of the initial kinetic energy Ek of the sputtered S atom,when the atom distance d=3 Å.
高速时(图4(b),S 原子的初始动能为30.4 eV),能量高的能级在交叉前(此时能级的主要成分为脱离晶格的S 原子的pz轨道)电子占据数为0,同样在d=1.8 Å附近出现电子占据,当d> 2.4 Å后(此时能级的主要成分为脱离晶格的S 原子的pxy轨道)电子占据数为1.82.能级交叉前低能量的pxy轨道是满占据,因此大约有0.18 个电子从pxy轨道跃迁至pz轨道.
能级交叉后,能量高的能级上的电子占据数随S 原子初始动能的变化如图4(c)所示.随着S 原子的初始动能增大,能量高的能级上的电子占据数增加,即非绝热效应增强,这也与LZ 模型相符.由于电子占据了能量高的能级,推高了系统的能量,因此非绝热效应导致S 原子脱离晶格的势垒上升,即S 原子空位形成所需的能量增加,这就解释了图2(b)中势垒随S 原子的初始动能增加而增加的趋势.
除能级上的电子占据数外,有电子占据的能级的能量也会影响S 原子脱离晶格的能量势垒.比较图3(b)和图3(c),注意到在低速和高速两种情况下,能级交叉后其能量的走势也不尽相同.在低速情况下,如图3(b)所示,虽然各能级的能量出现一定程度的振荡,但总的趋势是在能级交叉后其能量降低;在高速情况下,如图3(c)所示,在能级交叉后,电子主要占据的能级则能量升高.特别指出,虽然如上所述其中一条pxy轨道能级不与其他能级耦合,但由于这条能级上一直是满占据,因此该能级的能量走势仍对S 原子脱离晶格的势垒有重要影响: 当该能级的能量降低时,S 原子脱离晶格的势垒也较低,而当该能级的能量升高时,势垒也将被推高.
能级能量走势的差异是由库仑相互作用导致.不参与耦合的能级主要由脱离晶格的S 原子的px和py轨道构成,与能量高的能级的轨道成分相同.当脱离晶格的S 原子初始动能低时,能量高的能级上的电子占据数小,该能级与不耦合的能级之间的库仑排斥作用弱,因此这两条能级能量的走势下降.然而,当脱离晶格的S 原子初始动能高时,能量高的能级上的电子占据数大,如图4(c)所示甚至超过1.5,由于该能级和不耦合的能级轨道成分相同,能级之间的库仑排斥作用强,因此两条能级的能量都被推高.发现当S 原子初始动能小于20.9 eV 时,能级交叉后其能量普遍下降,而初始动能大于22.8 eV 时,能级交叉后其能量普遍上升,即在20.9—22.8 eV 附近存在转变.这一转变点与图2(b)中势垒的阶跃式增长所对应的S 原子的初始动能一致.
根据以上讨论可知,当脱离晶格的S 原子初始动能Ek较低时,即在7.6—20.9 eV 之间时,随着S原子的初始动能增加,非绝热效应增强,更多电子占据能量高的能级(LZ 模型),因此S 原子脱离晶格的势垒线性增加.当Ek从20.9 eV 升至22.8 eV时,能量高的能级上的电子占据数迅速上升,且能级交叉后电子占据的能级能量走势由下降转变为上升,因此能量势垒呈现出阶跃式的增长.当Ek超过22.8 eV 后,势垒随初始动能仍线性增长,其增长的斜率与Ek低于20.9 eV 时相近,即在高速区势垒的增长仍由高能量能级的电子占据数增多导致.由此可见,图2(b)中S 原子脱离晶格的势垒由LZ 电子跃迁和能级间的库仑排斥共同决定.
3.3 电荷分布
下面讨论非绝热效应对S 原子脱离晶格过程中电荷分布的影响.图5 给出了脱离晶格的S 原子在不同初始动能情况下,运动至距晶格不同位置时,非绝热模拟与绝热模拟所得的电荷密度之差.

图5 当脱离晶格原子的运动距离d 取不同值时,非绝热模拟与绝热模拟所得的电荷密度之差(蓝色和黄色等值面分别代表电荷的减少和增加) (a)—(c) 低速条件(Ek=15.2 eV)下,(a) d=1.75 Å,(b) d=2.25 Å,(c) d=5.00 Å;(d)—(f) 高速条件(Ek=30.4 eV)下,(d) d=1.75 Å,(e) d=2.25 Å,(f) d=5.00 ÅFig.5.Charge density difference between the non-adiabatic and adiabatic results at different atom distances d (Blue and yellow isosurfaces represent the charge depletion and accumulation respectively): (a)—(c) When the initial kinetic energy of the sputtered S atom is low (Ek=15.2 eV),(a) d=1.75 Å,(b) d=2.25 Å,(c) d=5.00 Å;(d)—(f) when the initial kinetic energy of the sputtered S atom is high (Ek=30.4 eV),(d) d=1.75 Å,(e) d=2.25 Å,(f) d=5.00 Å.
当d=1.75 Å时,如图5(a)和图5(d)所示,非绝热模拟与绝热模拟所得的电荷密度分布相同.这是由于此时系统能级尚未发生交叉,电子完全处于基态,不存在非绝热效应.当d=2.25 Å时,高能量的能级上出现了电子占据,非绝热效应已然显现.原子速度较低时,如图5(b)所示,非绝热模拟中电荷更多地出现在脱离晶格的S 原子的pz轨道上.这是因为如前所述,在低速情况下,能级交叉后大量电子从pxy轨道跃迁到pz轨道.当原子速度较高时,如图5(e)所示,非绝热模拟中电荷则更多是出现在S 原子的px与py轨道上.这也是因为如前所述,在高速情况下,能级交叉后pz轨道上几乎没有电子占据.这些结果表明非绝热效应影响了脱离晶格的S 原子上电荷的轨道分布.此外,非绝热效应也导致MoS2晶格内电荷分布的改变,且高速时这种改变更明显.当d=5.00 Å时,S 原子完全脱离了晶格,空位缺陷已经形成.脱离晶格的S 原子上的电荷分布情况与d=2.25 Å时的类似,即在低速情况下,其pz轨道上电荷更多,而在高速情况下,其pxy轨道上电荷更多.还可以看到,不论原子初始动能如何,MoS2晶格中的电荷都减少,即S 原子脱离晶格的过程中从晶格内多带走了一部分电荷.
为更清楚地了解脱离晶格S 原子上的电荷变化情况,利用Hirshfield 算法[36]计算了该原子上的电荷数,其结果如图6 所示.随着脱离晶格的S 原子逐渐远离MoS2,该原子上的电荷数不断增加.当S 原子的运动距离d< 1.8 Å时,能级尚未交叉,系统内不存在非绝热效应,因此脱离晶格的S 原子所携带的电荷数目不依赖于其初始动能.当d> 1.8 Å时,电子跃迁开始发生,图6 中不同初始动能所对于的曲线开始展现出差异.无论初始动能多大,脱离晶格的S 原子上的电荷数都大于6,即从晶格中带出了额外的电荷.其基本趋势是随着S 原子的初始动能增大,非绝热效应变强,S 原子带出的电荷数也增大,即在S 原子空位缺陷附近留下了更多的空穴.这说明非绝热效应也改变了晶格中缺陷处的电荷分布.
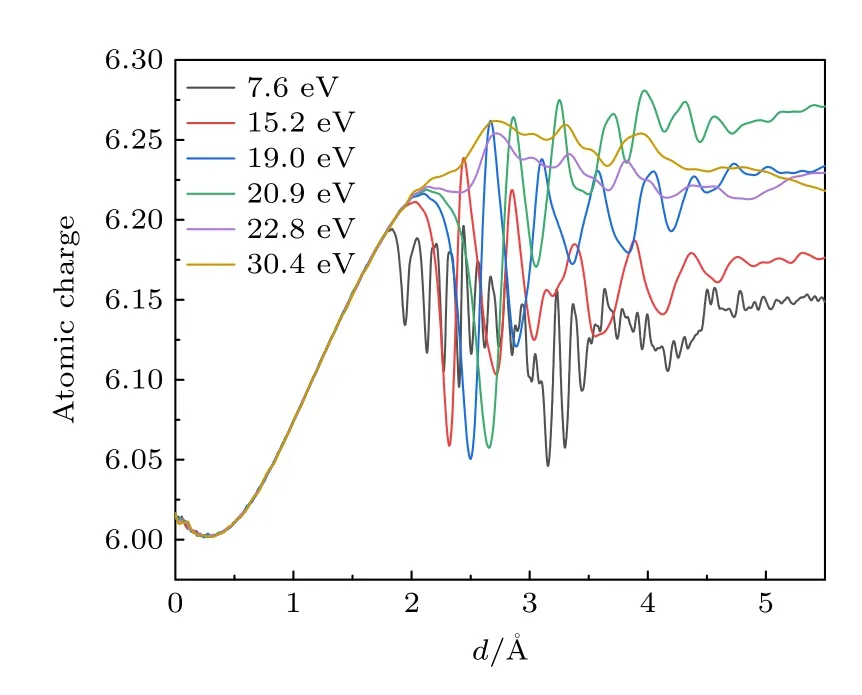
图6 不同初始动能下,非绝热模拟中脱离晶格的S 原子上的电荷数随d 的关系Fig.6.Number of charges on the sputtered S atom as a function of d with different initial kinetic energies.
4 结论
采用TDDFT 模拟研究了MoS2系统中的S 原子脱离晶格形成空位缺陷过程中的电子动力学.S 原子脱离晶格时具有很大的初始动能,该过程是非绝热的,涉及到系统内的电子跃迁至激发态.非绝热效应的出现影响了S 原子脱离晶格的能量势垒.相比于绝热近似,考虑非绝热效应后S 原子需消耗更多能量以脱离晶格的束缚.随着S 原子的初始动能增大,更多电子被激发到高能量的能级,即非绝热效应变强.当S 原子的初始动能大于22 eV后,由于大量电子占据了高能量的pxy能级,库仑排斥作用使得pxy能级的能量进一步上升.电子的非绝热跃迁和库仑作用共同导致了S 原子脱离晶格的能量势垒随初始动能增加而增加,并在Ek=22 eV 附近发生阶跃式增长.此外,非绝热效应还使得S 原子脱离晶格时携带了更多的电子,即在晶格中空位缺陷附近留下空穴.在低速情况下,脱离晶格的S 原子上电荷更多地分布在pz轨道,而在高速情况下,电荷则更多地分布在pxy轨道.值得指出的是,尽管在S 原子脱离晶格的过程中系统并未产生磁矩,但SOC 仍然对系统性质有不可忽略的影响,因此必须被考虑.
利用非绝热模拟,考虑系统中电子跃迁带来的影响,对MoS2系统中的S 原子空位形成过程有了更清晰、深入的认识.相比于绝热模拟,非绝热的模拟结果更加接近真实的实验情形,也能更准确地反映材料理化性质的变化过程.除S 原子空位外,MoS2材料其他缺陷的形成中也可能涉及到非绝热过程,因此也可以利用TDDFT 方法研究其他缺陷形成过程中的电子动力学.此外,由于其他过渡金属硫属化合物与MoS2有相似的物理性质,我们预期在这些材料中能观察到相似的电子非绝热动力学行为.
