永登七山羊全基因组选择信号检测分析
2023-11-30栗登攀马克岩韩金涛白雅琴李讨讨马友记
栗登攀,马克岩,韩金涛,白雅琴,李讨讨,马友记*
(1.甘肃农业大学动物科学技术学院,兰州 730070;2.永登县动物疫病预防控制中心,兰州 730300; 3.甘肃省畜牧技术推广总站,兰州 730030)
我国地方绵羊品种资源丰富,为切实挖掘新资源并加以利用,在全国第三次畜禽资源普查中发现了永登七山羊。永登七山羊在永登县七山乡各村落均有分布,该群体具有特征一致、特性明显、遗传性能稳定等特点。研究发现,永登七山羊与兰州大尾羊、滩羊和岷县黑裘皮羊之间分化明显,独立形成聚类[1]。
在动物漫长的驯化过程中,动物的行为习惯、体型外貌因受到自然选择而不断发生着巨大的变化[2-3]。使用选择信号来进行动植物的适应性进化研究越来越多[4-6]。选择信号是指动物在进化过程中受长期的自然选择和强烈的人工选择两种作用而遗留在基因组上的遗传标记,从基因组水平探讨种群间的遗传差异,有助于理解动物对选择适应的遗传机制和挖掘重要经济性状的候选基因,为种质资源的合理利用、保存提供理论依据[7-8]。Pan等[9]通过对89个中国地方绵羊品种进行全基因重测序揭示了RXFP2基因与角的独特形状相关。Li等[10]通过选择多种尾型的品种羊进行选择信号扫描,利用窗口XP-CLR和π ratio的方法,筛选出一系列(SGCZ、GLIS3、FGF7、PAPPA2、MAP2K3等)与脂肪沉积与代谢相关的基因。Li等[11]收集分析了中国、巴基斯坦和尼泊尔的15个代表性家山羊品种共115个样本以及4个野山羊样本的全基因组数据,筛选出调控山羊热适应、产绒和产奶的关键候选基因与SNP错义位点。这些研究结果有助于人们深刻了解动物受自然选择和人工选择的影响,并为物种起源及驯化机制提供见解。目前,对于永登七山羊的研究仅存在于遗传多样性分析上,需要进一步从基因组水平筛选鉴定可能与永登七山羊重要经济性状相关的功能基因。
本研究利用SLAF技术检测4种绵羊群体全基因组范围内的SNPs。通过主成分分析、基因流事件以及候选基因挖掘等方法,旨在从分子水平证明永登七山羊群体与其他群体的基因交流情况,并对适应性进化过程中的优势基因进行挖掘,从而为永登七山羊的保护利用提供重要的理论参考。
1 材料与方法
1.1 试验材料
本研究基于前期针对地方品种(永登七山羊)的遗传多样性分析研究[1],选择兰州大尾羊(LFT)、滩羊(TAN)、岷县黑裘皮羊(MBF)作为参考对象。如表1所示,每个品种采集10只健康的成年母羊,颈部静脉采血3~5 mL,用5 mL EDTA抗凝管收集,置于-80 ℃冰箱保存备用。

表1 样品信息Table 1 Sample information
1.2 试验方法
对抗凝血样提取DNA,利用NanoDrop 2000和Qubit 2.0检测DNA的纯度和浓度。采用琼脂糖凝胶电泳检测基因组 DNA 的完整性。对DNA质检合格的样品采用双酶切的RAD建库方式,将长度范围414~464 bp的酶切片段定义为SLAF标签,对SLAF标签进行处理后在Illumina HiSeqTM 2500测序仪上进行简化基因组测序。测序由北京百迈客生物科技有限公司完成。
1.3 数据处理与分析
对测序得到的原始数据按照酶切效率、测序质量和片段选择进行数据质量评估,得到各个样品的原始序列片段。其中限制性内切酶组合为Hpy166 II+EcoR V,采用读长126 bp×2 作为后续的数据评估和分析数据。利用bwa[12]将原始数据比对到绵羊参考基因组(oar_v4.0)上,使用GATK[13]和samtools[14]两种方法开发 SNP,取SNP标记交集并使用snpeff软件[15]用于注释变异。
1.4 主成分分析
使用EIGENSOFT[16]软件包中的smart PCA程序基于SNP数据,在PCA二维聚类图[1]基础上重新进行数据分析,得到4个绵羊群体的三维聚类情况。
1.5 基因流分析
利用Treemix软件,使用全基因组范围内等位基因频率数据,推断多个群体分裂和混群模式。使用R的OptM判断最优m值,m值取1~10。每个m值重复5次后,使用最优m值进行 Treemix 分析,绘制群体最大似然树;并在最大似然树上用箭头展示渗入的迁移事件。
1.6 选择清除分析
基于SNP数据,按照次要等位基因频率(MAF:0.05)和位点完整度(INT:0.8)进行过滤,获得高一致性SNP位点,按照100 kb的窗口、10 kb的步长对染色体进行选择性清除区域检测。使用PopGenome软件包计算群体分化指数(Fst)、核苷酸多态性(π)和多样性变化倍数(θπ ratio)等群体遗传学指标。在筛选选择性清除区域时,分别以Fst和π ratio前5%分位数对应的数据作为阈值,取该区域的交集为选择性清除区域。
1.7 受选择区域筛选及基因富集分析
针对选择性清除区域进行相邻区域窗口合并,统计合并后窗口的位置信息以及窗口内基因数目,对应的基因位置。使用Enesmbl数据库的BioMart功能,利用功能分类和代谢途径信息对候选基因进行基因功能注释。利用DAVID数据库对基因进行 GO(Gene Ontology)和 KEGG(Kyoto Encyclopedia of Genes and Genomes)富集分析,并分别生成富集分析图表。
2 结 果
2.1 SNP检测
本项目共获得 709 067 个SLAF标签,标签的平均测序深度为 17.63x,其中,多态性SLAF标签有 445 768 个,共获得 1 658 596 个SNPs标记。根据SLAF在染色体上的分布,绘制SLAF在染色体上的分布图(图1)。
2.2 主成分分析
根据主成分分析推断出群体之间聚类模式的差异。由4个绵羊群体的主成分分析(图2)可明显看出,PC1、PC2和PC3的贡献率分别为3.86%、3.43%和3.39%;LFT群个体最为聚集,MBF群体最为分散;MBF、QS和TAN群个体可独立分群。
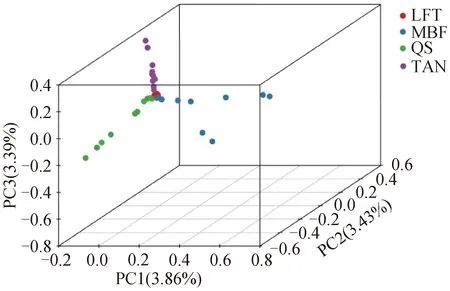
图2 4个绵羊群体的主成分分析Fig.2 Principal component analysis of 4 sheep populations
2.3 基因流分析
基因流动是指位于不同种群之间或者在种群内部产生的遗传物质的交流[17-18],既能抵消由遗传变异和自然选择引起的自我种群之间的分化,又能给整个大的群体中带来新的变异。从图3可以看出,TAN和LFT群体间存在较强的基因交流,QS和LFT群体间发生较弱的基因交流。
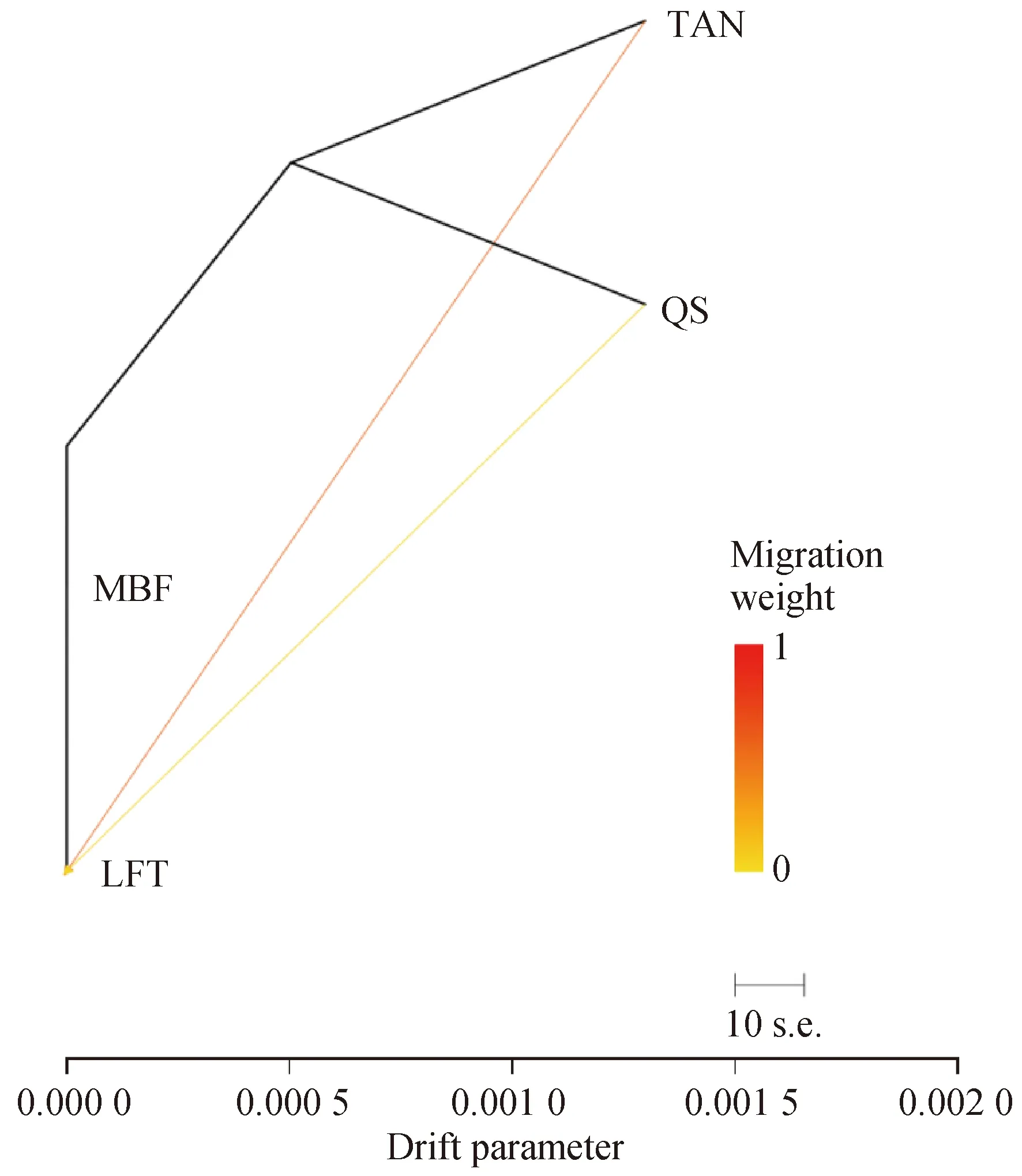
图3 基因流动Fig.3 Gene flow
2.4 群体间受选择区域筛选
将QS与LFT、TAN和MBF进行两两比较并将其余3个群体数据合为整体与QS进行比较。在QS与MBF之间,将Fst>0.273, π ratio>8.55的基因组交集区域定义为QS与MBF群体间选择信号(图4A),检测到163个受选择窗口,注释到424个候选基因(图5A);以同样的方法,在QS与LFT之间筛选到127个受选择窗口(图4B),注释到294个候选基因(图5B);在QS与TAN之间筛选到133个受选择窗口(图4C),注释到301个候选基因(图5C)。
在QS与(MBF +LFT+TAN)(简称:MLT)之间,筛选到202个受选择窗口,并绘制群体间选择信号示意图(图4D);注释到466个候选基因(图5D)。另外,统计窗口中各个染色体的Fst和π ratio值,以及窗口内各个基因的位置,绘制QS与MLT SNPs注释表(表2)。
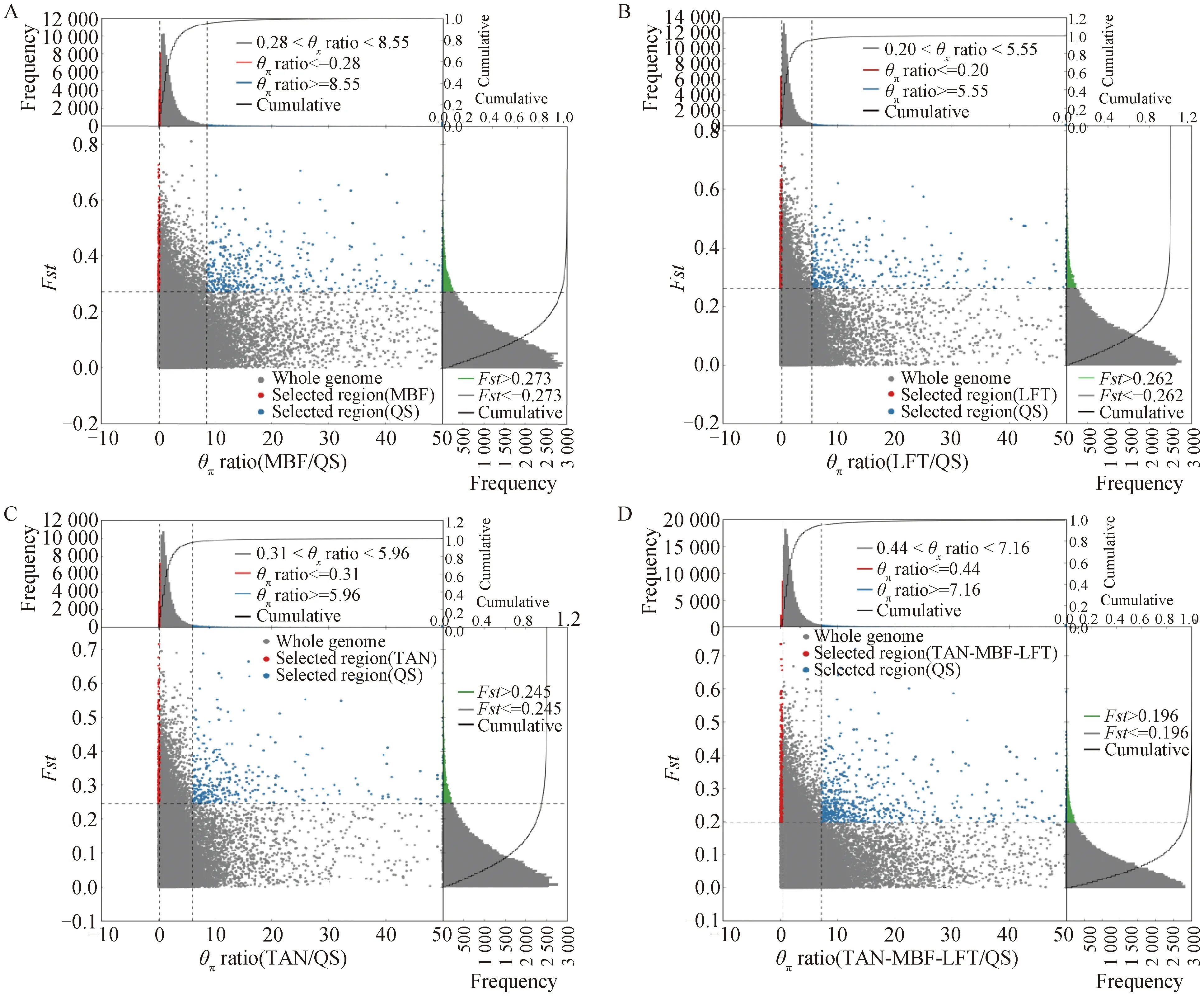
A.QS与MBF组;B.QS与LFT组;C.QS与TAN组;D.QS与MLT组 A.QS and MBF group;B.QS and LFT group;C.QS and TAN group;D.QS and MLT group图4 群体间选择信号示意图Fig.4 Schematic of selection signatures between populations
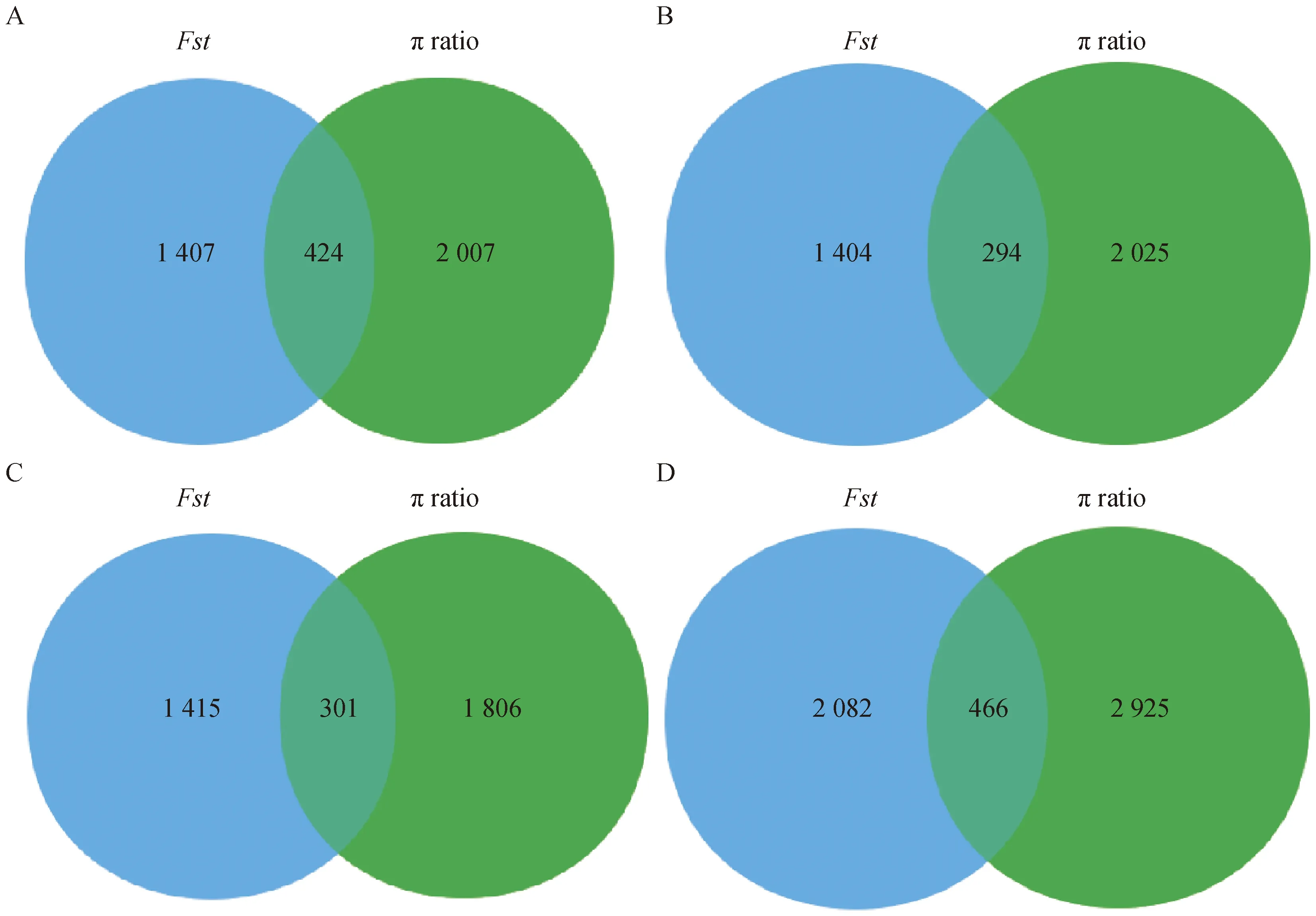
A.QS与MBF组;B.QS与LFT组;C.QS与TAN组;D.QS与MLT组A.QS and MBF group;B.QS and LFT group;C.QS and TAN group;D.QS and MLT group图5 两种方法检测的正选择基因韦恩图Fig.5 Venn diagram for the positively selected genes identified via two approaches
2.5 GO富集分析
对候选基因进行GO功能富集分析。在QS与MBF组间424个候选基因中,119个候选基因显著富集于65个GO条目(P<0.05),其中包括9条细胞组分类,43条生物过程类和13条分子功能类,主要集中在细胞极性的建立或维持、肺泡发育、组蛋白H3-K4三甲基化、血管重塑等。在QS与LFT组294个候选基因中,119个候选基因显著富集于79个GO条目(P<0.05),其中包括54条生物过程类,18条分子功能类和7条细胞组分类,主要富集在mRNA稳定化、前置/后置模式规范、有丝分裂期板块聚集等。在QS与TAN组301个候选基因中,90个基因显著富集于41个GO条目(P<0.05),其中包括33条生物过程类和8条分子功能类,主要富集在烟酸和烟酰胺的代谢、金黄色葡萄球菌感染、雌性激素信号传导途径、初级胆汁酸的生物合成等。
另外,在QS与MLT组466个候选基因中,130个候选基因显著富集在124个GO条目(P<0.05),其中包括96条生物过程类,14条细胞组分类和14条分子功能类,主要富集在多细胞进程、血液循环、发育生长调节、钙离子转运的正调节等过程。富集条目、通路名称、P值以及对应基因见表3。
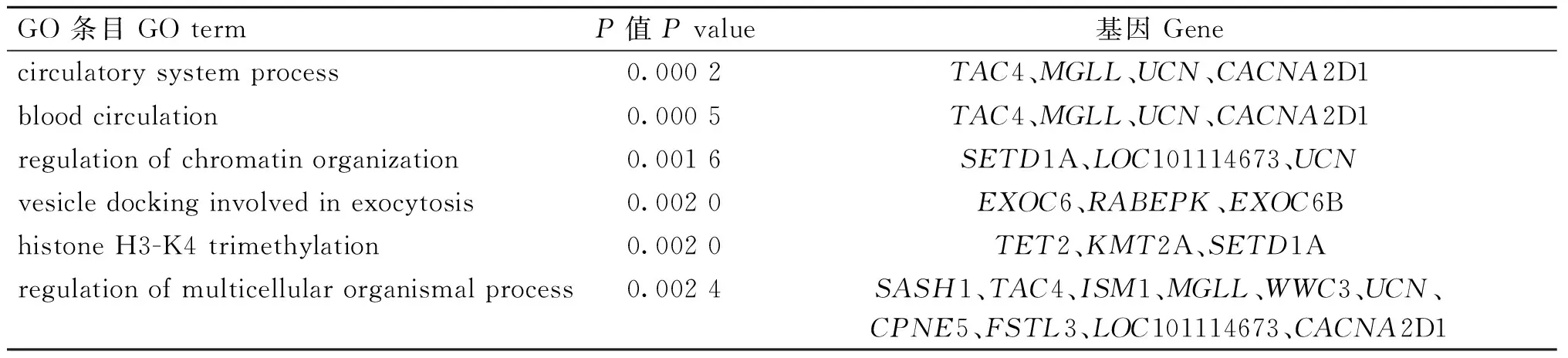
表3 GO功能富集分析Table 3 GO function enrichment analysis
2.6 KEGG富集分析
对候选基因进行KEGG通路富集分析,并挑选前10个显著的KEGG通路作富集气泡图(图6)。结果显示,QS群体与MBF、LFT、TAN品种相比较,分别有231条、222条和214条信号通路参与主要生化代谢途径和信号转导途径。其中,QS与MBF比较组中显著富集通路有15条,其中15个候选基因(GAD1、CYP11B1、PTAFR、CAMK2D、CDH1、BMP2等)可能涉及重要经济性状,主要参与类固醇激素生物合成、叶酸的生物合成、雌激素信号传导途径、金黄色葡萄球菌感染、卵巢类固醇生成、花生四烯酸代谢、精氨酸和脯氨酸的代谢等通路。QS与LFT比较组中显著富集通路有22条,涉及12个候选基因(ALDH1A1、GAD1、GRM1、PTAFR、CYP11B1等),主要集中在雌性激素信号传导途径、金黄色葡萄球菌感染、视黄醇代谢、丙氨酸、天门冬氨酸和谷氨酸的代谢、苯丙氨酸的代谢、色氨酸的代谢、类固醇激素的生物合成等。QS与TAN比较组中显著富集通路有10条,涉及13个候选基因(为ALDH1A1、KMT2A、PARD6G、SETD1A、CAMK2D、GRM1等),主要集中在烟酸和烟酰胺的代谢、金黄色葡萄球菌感染、雌性激素信号传导途径、初级胆汁酸的生物合成、氮素代谢、粘着剂的交界处、赖氨酸的降解、视黄醇代谢、TGF-beta信号传导途径、轴突引导、嘧啶的代谢等。
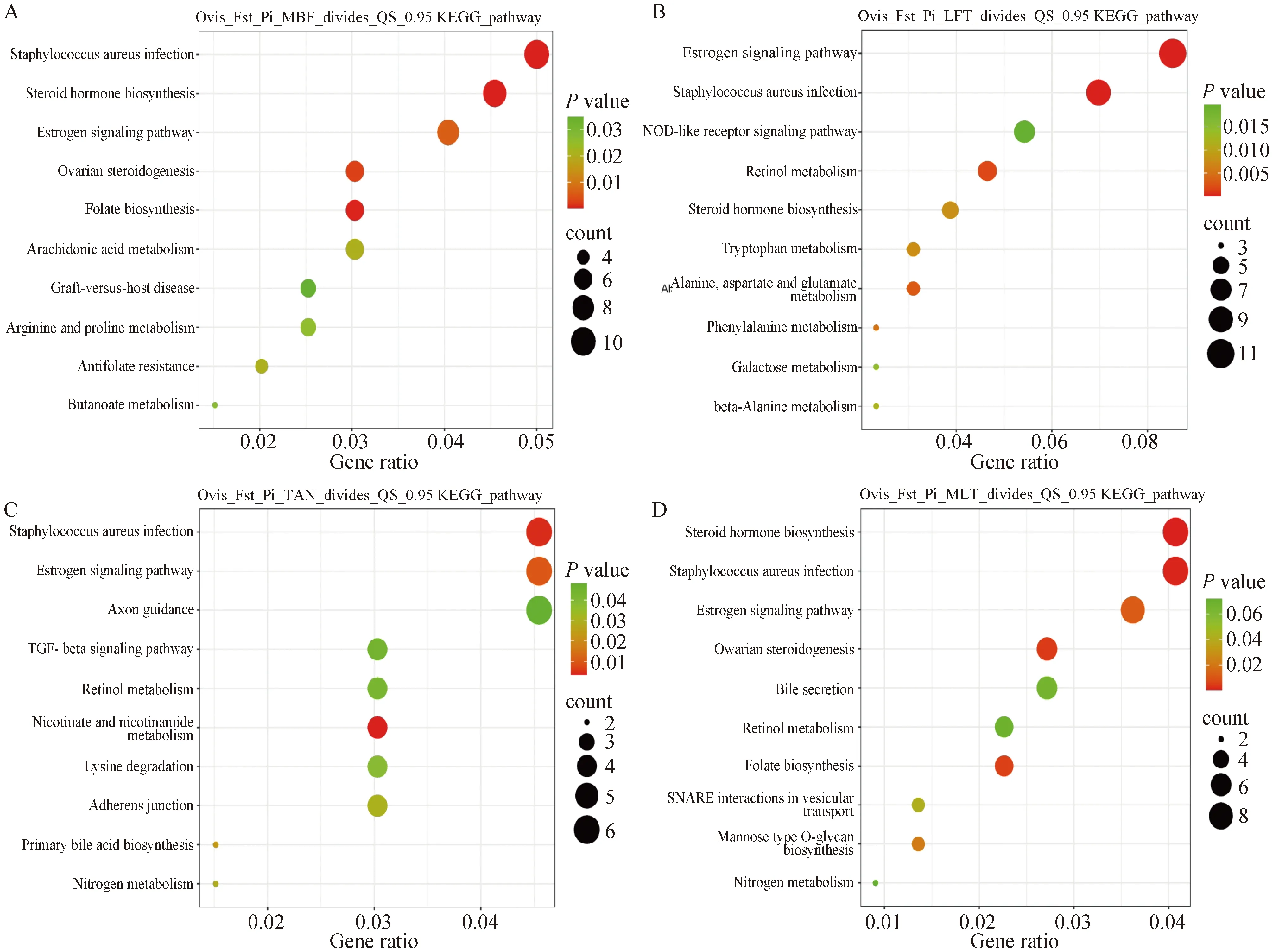
A.QS vs MBF组;B.QS vs LFT组;C.QS vs TAN组;D.QS vs MLT组A.QS and MBF group;B.QS and LFT group;C.QS and TAN group;D.QS and MLT group图6 KEGG 通路分析Fig.6 KEGG pathway analysis
QS与MLT比较组中,参与主要生化代谢途径和信号转导途径有244条。其中PTAFR、UGT2A1、KRT15等基因主要参与类固醇激素生物合成、叶酸合成、雌激素信号通路、金黄色葡萄球菌感染、卵巢类固醇生成、甘露糖型O-聚糖生物合成、SNARE在膀胱运输中的相互作用等。
值得注意的是,雌激素信号通路和金黄色葡萄球菌感染通路均显著存在于4个组别中,并从中均发现V15、K38、KRT32、KRT35、KRT36这5个基因。
3 讨 论
在动物遗传学上,基于不同群体的SNP差异程度,可将各群体进行分群。本研究中,通过PCA分析,发现岷县黑裘皮羊、永登七山羊和滩羊均可与其他群体分开,除永登七山羊外,其他群体与之前一些学者的研究结果一致[19-20]。
基因交流可以向其他群体中引入新的等位基因,进而影响群体遗传多样性,产生新的性状组合,发生遗传变异[21-23]。本研究发现,滩羊和兰州大尾羊之间发生过相对较强的基因交流,永登七山羊与兰州大尾羊之间发生了较弱的基因交流。其原因是滩羊、兰州大尾羊、永登七山羊均属蒙古羊,虽然永登七山羊饲养环境相对封闭,但因为和兰州大尾羊地理上接近,难免发生基因交流;也是自然选择作用的表现。因此保护这些品种生物多样性具有很大难度,可以采用等量留种的方式,尽量保持现有等位基因,防止近交衰退。
本研究取top 5%Fst和π ratio的交集用来确定基因组受选择区域,并对筛选出来的候选基因进行GO和KEGG富集分析。采用Fst和π ratio的交集来进行筛选鉴定,能够得到更加可靠的结果[24-25]。本试验中,各比较组的受选择窗口具有一定差异,且注释到的基因均随着受选择窗口的数量而变化。通过SNPs注释,发现了可能与永登七山羊重要经济性状相关功能的20个候选基因,包括CDH1、V15、K38、KRT15、KRT32、KRT35、KRT36、BMP2、GRM1、ALDH1A1、CAMK2D、MASP1、LUC7L3、UGT2A1、PTAFR、PLOD1、SETD1A、PARD6G、KMT2A、SOD1。其中与绵羊生长发育有关的功能基因有BMP2、GRM1和ALDH1A1。在MBF与QS比较组中筛选出了BMP2基因,发现BMP2基因存在于Hippo信号通路、基底细胞癌、转化生长因子-β(TGF-β)和细胞因子-细胞因子受体相互作用的信号通路中。BMP2基因影响羊生长性状和脂肪酸组成[26-27],在肌肉发育中发挥重要作用[28]。与脂肪尾表型,脂质代谢相关,参与了羊尾脂肪沉积[29-31]。除此之外,BMP2可能是影响绵羊产仔数的候选基因[32]。在滩羊与永登七山羊比较组和兰州大尾羊与永登七山羊比较组均筛选出GRM1和ALDH1A1基因。发现GRM1基因存在于雌激素信号通路、磷脂酶D信号通路、逆行内源性大麻素信号、长时程增强、长时程抑制、间隙连接的信号通路,GRM1基因被认为是影响断奶后增益的重要候选基因,对绵羊生长和肉类生产性状有一定影响[33-35],可以作为绵羊肉质性状的候选基因[36];又发现ALDH1A1基因存在于视黄醛代谢信号通路。ALDH1A1基因是绵羊脂尾进化关键基因[37],可能参与滩羊脂肪细胞分化调控,敲除滩羊尾部脂肪前体细胞中ALDH1A1基因能够抑制脂肪细胞分化过程[38]。
另外,雌激素信号通路均显著存在于4个组别中。雌激素介导的oar-miR-485-5p靶向PPP1R13B调控绵羊成肌细胞增殖,参与炎症反应、新陈代谢和干细胞存活[39-40]。雌激素与雌激素受体结合后可以调节细胞核中的基因转录,或者激活细胞质中的激酶进而发挥其作用[41],且雌激素介导的信号通路与毛囊周期性生长和发育有关[42]。V15、K38、KRT32、KRT35、KRT36均作为角蛋白,广泛存在于绵羊毛发中[43-44]。其中KRT36是影响羊毛弯曲性状的潜在候选基因[45]。在毛球下部至中部,KRT32和KRT35基因在角质层和纤维皮质层均有表达[46]。KRT32在毛球中部至角质生长层皮质中广泛表达,KRT35基因和毛囊透明蛋白参与初始分化的皮质层结构的建立,最终导致纤维弯曲[47-48]。本研究采用简化基因组测序检测永登七山羊选择信号,注释到大量候选基因。在候选基因功能显著性富集分析中,功能基因揭示了永登七山羊在尾部脂肪沉积、肌肉发育和毛发生长等方面受到了选择。
4 结 论
本研究通过主成分分析和基因交流揭示4个群体的进化关系,结果表明,永登七山羊群体与其他3个群体分化明显,且与兰州大尾羊发生基因交流;此外还筛选到与绵羊生长发育有关的功能基因BMP2、GRM1和ALDH1A1等。结果为挖掘永登七山羊生长发育等相关基因提供参考,为其遗传资源保护与开发利用提供科学依据。
