固体进样-发射光谱法测定钼矿石矿粉中的高含量钼
2023-11-27郭心玮李昆郝志红姚建贞徐进力白金峰
郭心玮,李昆,郝志红,姚建贞,徐进力,白金峰*
(1.中国地质科学院地球物理地球化学勘查研究所,河北 廊坊 065000;2.联合国教科文组织全球尺度地球化学国际研究中心,河北 廊坊 065000)
矿产资源供给是中国社会经济可持续发展所面临的重大课题。《全国矿产资源规划(2016—2020 年)》将钼元素(Mo)列为特种金属矿产资源,是目前正在走进的工业4.0 时代的关键元素,被广泛应用于钢铁、航天和国防等工业领域[1-2]。中国工业化进程对钼资源的需求与日俱增,钼矿及钼元素储量的探明作为找矿战略行动中不可缺少的环节,对钼元素的地球化学样品分析测试提出了新的要求。因此,钼矿石中钼元素含量的现有分析方法体系亟待完善。
现阶段,液体进样技术占据了国内外分析粉末状地质样品中多种无机元素的主导地位,辅以简单的固体进样技术[3]。根据《钨矿石、钼矿石化学分析方法 第2 部分:钼量的测定》(GB/T 14352.2—2010)规定,将钼含量为0.01%~5%的试料通过碱熔融水浸提后,在硫酸介质中以铜盐催化形成可溶性橘红色硫氰酸钼络合物,以吸光度计算钼量。有色金属标准《钼精矿化学分析方法 钼量的测定 钼酸铅重量法》(YS/T 555.1—2009)中规定以钼酸铅重量法测定钼含量≥40%的钼精矿样品。除上述标准外,常用的钼量液体分析方法还有电感耦合等离子体发射光谱法(ICP-OES)[4-8]、电感耦合等离子体质谱法(ICP-MS)[9-14],这两种方法多先使用多酸体系或碱体系在高温条件下将待测样品消解后再进行分析,所得结果具有高精密度、高准确度和低检出限的优点,但线性范围较窄(如1.0~100.0μg/mL[5]),测定高含量钼时仍需稀释,导致工作步骤繁琐,大量酸碱的使用还易对环境造成污染。固体进样技术则避免了上述问题。X 射线荧光光谱法(XRF)多选用四硼酸锂和偏硼酸锂作熔剂,熔融制片分析后,以经验系数和理论α系数对其基体效应进行校正[15-17],可测范围为0.01%~5.17%[16]。但此方法消耗的大量熔剂不符合绿色化学理念。随着科技的进步,电子探针、扫描电子显微镜、X 射线能谱的联用技术已可实现矿物组成等特征参数的自动化定量分析,但其仪器相对昂贵,且XRF 法分辨率低,X 射线峰背比低,分析灵敏度较低,定量分析精度低,可能出现不平行样品分析[18-21]。
相较于上述诸方法,无需使用酸碱的交流电弧原子发射光谱法(Arc-AES)进样量大幅减少,仅需将样品与光谱缓冲剂混合均匀后即可上机测试,更符合绿色化学的理念,目前已成为多目标地球化学填图76 种指标配套分析方案中,测定地球化学样品硼、锡、银、钼、铅的推荐方法[22-23]。本文针对钼矿石钼矿粉中的高含量钼元素的测定,采用交流电弧原子发射光谱法,以国家一级标准物质和国家一级合成硅酸盐光谱分析标准物质为标准曲线,以国家一级矿石标准物质为样品,通过对内标元素种类及谱线的选择、钼元素谱线的选择、电流加载程序的优化、摄谱时间的设定等条件进行实验,建立了交流电弧全谱直读原子发射光谱法测定钼矿石钼矿粉中的高含量钼分析方法。
1 实验部分
1.1 仪器、材料和工作条件
DC/AC Arc AES-8000 型全谱交直流电弧发射光谱仪(北京北分瑞丽分析仪器有限公司):光源为WJD 型交直流电弧发生器,可35℃恒温,内设2400条/mm 光栅刻线,色散率为0.65nm/mm,可测量230~340nm 波长范围内发射光谱。
电极为光谱纯石墨电极:上电极为直径4.0mm的柱状电极,下电极孔径×孔深×壁厚分别为3.8mm×4.0mm×0.6mm,颈部直径×颈长为2.6mm×4.0mm 的细颈杯状电极。
XZJ-54 振动搅拌仪(武汉探矿机械厂):频率范围为100~4000Hz。
保鲜膜:产品标准GB/T 10457,以聚乙烯(PE)为原料,氧气透过率(1.19×10-5±40%)mol/(m2·s),二氧化碳透过率(5.26×10-5±40%)mol/(m2·s),透湿量(39±40%)g/(m2·d)。
1.2 试剂与样品配制
光谱缓冲剂:由中国地质科学院地球物理地球化学勘查研究所研制,由质量比为22∶20∶44∶14的焦硫酸钾、氟化钠、三氧化二铝、碳粉组成,其中含有质量分数为0.007%的二氧化锗作为内标物质。
合成硅酸盐分析标准物质基物:由中国地质科学院地球物理地球化学勘查研究所研制,由质量比为72.0∶15.0∶4.0∶4.0∶2.5∶2.5 的二氧化硅、三氧化二铝、三氧化二铁、纯白云石、硫酸钠和硫酸钾组成。
2%蔗糖-乙醇溶液:将10g 蔗糖(中国医药集团有限公司,分析纯)溶于250mL 去离子水中后,加入250mL 无水乙醇(中国医药集团有限公司,分析纯),混合均匀。
国家一级矿石标准物质:GBW07141、GBW07142、GBW07143、GBW07144。国家一级合成硅酸盐光谱分析标准物质:GBW07711。国家一级合成硅酸盐光谱分析标准物质基体:GSES Ⅰ。
1.3 实验测试分析步骤
按比例要求称取粒径小于0.074mm 的国家一级标准物质与光谱缓冲剂于5mL 坩埚中,放入小玛瑙球,以保鲜膜覆盖坩埚口,置于XZJ-54 振动搅拌仪上以2400Hz 研磨30min,混匀后的样品装入下电极中,滴入两滴质量分数为2%的蔗糖-乙醇溶液,置于70℃烘箱中干燥1h。
将样品置于交流电弧直读原子发射光谱仪上,用垂直电极法进行摄谱,分析元素背景谱线和内标元素背景谱线由原子发射光谱仪自动扣除,采用内标法以对数坐标二次曲线拟合,以标准物质的测量值与标准值的对数偏差(△lgC)表示准确度,对数偏差在-0.1~0.1 间数据符合实验要求。
2 结果与讨论
2.1 内标元素与分析线对的选择
内标元素的加入可减少由于激发电源电压不稳、基体成分所带来的干扰,保证良好的重现性和分析的灵敏度、准确度。内标元素多采用与不会对被测元素产生较大干扰且与其蒸发行为趋于一致的物质[24]。在交流电弧发射光谱法分析地球化学样品的主流分析方法中,因锗元素在地球化学样品中含量基本稳定在1~2μg/g,且其蒸发过程与目标元素钼较为接近,因此多选锗[25-26]元素作为内标元素,此外还有铕元素[27]及钯元素[28]等。本研究加入约为样品所含锗元素的50 倍以上的二氧化锗作为内标,以保证内标元素含量的一致性。
AES-8000 仪器中,Mo 的推荐分析线有14 条,考虑到钼矿石钼矿粉中的钼含量较高,本研究优先选择灵敏度较弱的Mo 分析线。Ge 的两条分析线有2 条,分别落于326.9494nm 和270.9626nm 处。综合考虑灵敏度和干扰信息后,以GBW07142 为样品,通过蒸发曲线对比两元素分析谱线间关系(图1),发现Mo 277.54nm 分析线与Ge 326.9494nm 分析线的蒸发行为较为一致,在10~30s 内信号强度增减基本保持一致,30s 后两个元素基本蒸发完全。因此,本法选择灵敏度相对较弱的Mo 线(277.54nm)作为测定高含量钼的分析线,选择Ge 线(326.9494nm)作为内标线,使得分析线和内标线对既可以保持相对一致的蒸发行为,又互不干扰。背景位置由仪器自动计算并扣除。
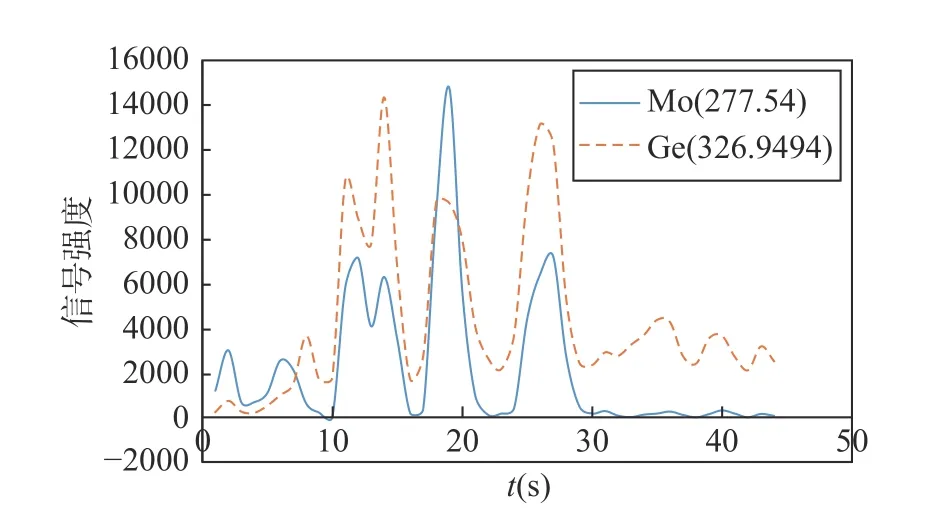
图1 Mo(277.54nm)和Ge(326.9494nm)蒸发行为比较Fig.1 Comparison of evaporation behaviour of Mo(277.54nm)and Ge(326.9494nm).
2.2 样品与缓冲剂比例的选择
光谱缓冲剂的加入可以有效地降低基体成分对弧焰温度的影响,增强待测元素的分馏强度、稳定弧焰、减弱光谱背景,以此制造对被测元素较为有利的激发条件,同时使试样基体更为趋同,进一步降低基体效应和背景[29]从而降低检出限,并获得更好的准确度与精密度[30-31]。文献[29-33]中大多采用样品与缓冲剂质量比为1∶1 时测定地球化学样品中银、硼、锡、钼、铅含量;郭心玮等[34]实验了样品与缓冲剂质量比为1∶2 时地球化学样品中银、硼、锡、钼、铅含量;曲红静等[35]以1∶5 比例测定了钛白粉样品中的13 中杂质元素。
研究表明,缓冲剂中的焦硫酸钾可在激发过程中将钼转化为沸点较低的二氧化钼或三氧化钼[36],因此,适当增加缓冲剂的比例有助于分析钼矿石、钼矿粉中钼含量。本实验对比了样品与缓冲剂质量比为1∶1、1∶2、1∶3 和1∶4 时同一样品的测量结果:当样品与缓冲剂比例为1∶1 时,在激发过程中容易发生溅射,造成其检出限、重现性和准确度出现较大的误差;当样品与缓冲剂比例为1∶2 时,就已经有效地改善了蒸发行为;如果单纯地增加缓冲剂比例,虽可提高检测上限,但在实际样品测量过程中会因缺乏相似基体组分,使得数据可靠性降低。
2.3 电流程序的选择
电流程序将直接影响样品的蒸发行为。样品激发过程中,可首先设置强度较低、保持时间较短的一级电流。经试验表明,当一级电流小于3A 时,电极起弧效果不理想;4A 时即有显著改善。起弧后,应衔接强度较高、保持时间较长的二级电流,实现Mo元素测定具有较好的信噪比。试验测试了5A、10A、15A、20A、25A 二级电流强度。结果表明,当二级电流强度小于15A 时,信噪比随二级电流强度的增加而增大,但当二级电流强度大于15A 时,背景强度增强,信噪比降低。因此,实验二级电流选择15A。
在一定程度内,Mo 元素和Ge 元素信号强度随曝光时间增长而增强,曝光时间的选择将会影响待测元素各项指标及分析效率。实验以GBW07142 为样品,考察了截取曝光时间对样品强度的影响。图2以截取曝光时间为横坐标,以Mo 元素信号累加强度为纵坐标作图。从图中可以看出,当截取曝光时间在30s 前,Mo 元素和Ge 元素强度均显著增加;35s后,Mo 元素信号强度增加不显著。结合样品分析效率,选择截取曝光时间为35s。经综合分析,实验所用电流程序设置为:一级电流4A,保持时间5s,二级电流15A,保持时间30s,总截取曝光时间为35s。

图2 曝光时间选择Fig.2 Choice of exposure time.When the exposure time is less than 30s,the evaporation intensity of Mo and Ge increases significantly; however,after 35s,the evaporation intensity of Mo reaches a plateau.
2.4 标准曲线的绘制
固体原子发射光谱常用标准系列有合成硅酸盐标准物质系列和由不同性质、不同含量的国家一级标准物质所构成的天然地球化学标准物质系列[37]。本实验根据钼矿石特性,选择由国家一级合成硅酸盐光谱分析标准物质GBW07711 和国家一级矿石标准物质GBW07141、GBW07142、GBW07143、GBW07144 配制而成的标准系列。标准系列中各级标准按表1 的配比方案研磨待用,各级标准样品中钼含量列于表1。标准曲线选择Mo(277.54nm)为分析线,以Ge(326.9494nm)为内标线,电流程序设置为:一级电流4A,保持时间5s,二级电流15A,保持时间30s。采用内标法以对数坐标二次曲线拟合,所得标准曲线方程为y=-0.077x2+1.3077x+1.2725,决定系数(R2)达0.999。标准曲线范围为500~500800μg/g,比《钨矿石、钼矿石化学分析方法 第2 部分:钼量的测定》(GB/T 14352.2—2010)中检测上限(5%)高出百倍,可在不稀释情况下直接测定地球化学样品中高含量钼。
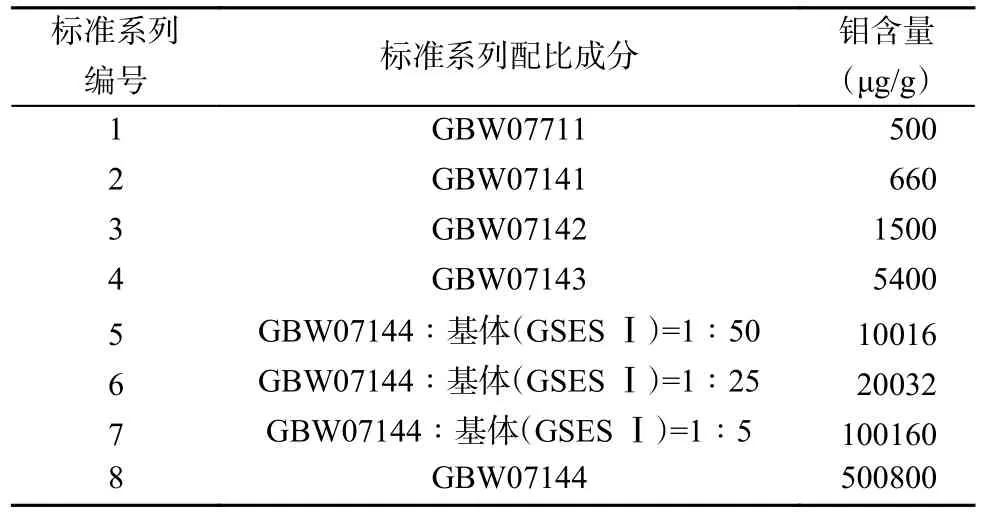
表1 标准系列配比及钼含量Table 1 Ratio of the standard series and concentration of Mo.
2.5 方法检出限
检出限是分析测试的重要指标,对于仪器性能的评价和方法的建立都是重要的基本参数之一。根据上述实验条件,以国家一级标准物质GBW07141和光谱缓冲剂1∶2 混合均匀,平行分析7 次,钼含量分别为:683.15、676.11、681.28、678.51、659.32、679.70、664.36μg/g,以计算结果的3 倍标准偏差(3σ)表示方法检出限,为27.38μg/g,略高于碱熔-电感耦合等离子体发射光谱法(0.002%)[9]和X 射线荧光光谱法(0.0026%)[16]测定钼矿石中钼含量的检出限。
2.6 方法准确度和精密度
实验选取国家一级标准物质GBW07141、GBW07142、GBW07143 进行精密度实验,每个样品按照所选方法平行分析测定12 次,以12 次分析结果的相对标准偏差(RSD)表示精密度,以12 次分析结果的相对误差和每个标准物质平均值与标准值的对数偏差(△lgC)表示准确度,结果如表2 所示。表中,以AVE 表示12 次分析结果平均值。从表中可以看出,RSD 在3.28%~8.30%,与标准值之间的相对误差为-0.43%~0.73%。本方法测定值与标准值基本吻合,具有较好的精密度和准确度,符合《多目标区域地球化学调查规范(1∶250000)》(DZ/T 0258—2014)中对检出限三倍以上,1%含量以下样品精密度和准确度的要求(△lgC≤0.05,RSD≤10%)。

表2 方法精密度和准确度Table 2 Accuracy and precision of the method.
2.7 方法对比
本方法检出限(27.38μg/g)与碱熔-电感耦合等离子体发射光谱法(0.002%)和XRF 法(0.0026%)基本持平,且可不用稀释直接测定钼矿石、钼矿粉中高含量钼,所得数据符合地质实验室分析要求。而且,本方法在更宽的测定范围内可得到准确可靠数据的同时,解决了ICP-MS 法、ICP-OES 法、滴定法、重量法和吸光度法中操作繁琐、试剂使用量大、污染大的问题,解决了XRF 压片法所需样品量大、存在辐射等问题,填补了发射光谱法测定高含量Mo 的方法空白,大幅提高了发射光谱法测定钼矿石、钼矿粉中元素Mo 的测定上限,实现Mo 元素百分含量达50%的精准分析。
3 结论
本研究提出将钼矿石样品与光谱缓冲剂以质量比1∶2 混合均匀后,以钼弱灵敏线(277.54nm)为分析线,以锗弱灵敏线(326.9494nm)为内标线,采用垂直电极法,一级电流以4A 强度保持5s,二级电流以15A 强度保持30s,采用自研标准系列作标准,由仪器自动扣除分析线和内标线背景后以对数坐标二次曲线拟合计算。方法测定范围为500~500800μg/g,检出限为27.38μg/g,相对标准偏差为3.28%~8.30%,相对误差为-0.43%~0.73%。建立了可不用稀释直接测定钼矿石、钼矿粉等地球化学样品中高含量钼样品的交流电弧发射光谱法。本方法在实现操作简便、环境友好的测试条件下,保持了现有方法精密度和准确度的同时,将高含量钼样品的分析上限从5%提高到50%。
值得注意的是,在实际应用中,由于钼矿石、钼矿粉中钼含量范围较大,且本方法所选用的弱灵敏线无法测定低含量钼,对于含量跨度较大的样品,无法实现一次测量。下一步将以本实验为基础,开发AES-8000 全谱交直流电弧发射光谱仪多线测量模式,选择不同灵敏度的波长来测量钼元素含量,实现高、中、低含量一次测量。
