正丙苯高温氧化机理的分子动力学模拟研究
2023-11-24周子豪王思皓黄玳川刘波甯红波
周子豪, 王思皓, 黄玳川, 刘波, 甯红波
正丙苯高温氧化机理的分子动力学模拟研究
周子豪, 王思皓, 黄玳川, 刘波, 甯红波
(西南交通大学材料先进技术教育部重点实验室, 成都 610031)
正丙苯是Jet A、 Jet A-1及国产RP-3航空煤油中芳香烃的典型替代组分. 本文采用基于反应力场的分子动力学模拟研究了正丙苯高温氧化过程的主要反应网络、 主要产物的形成机理以及在不同温度、 密度和当量比条件下正丙苯氧化主要产物的分布规律, 并结合反应动力学理论计算了正丙苯高温氧化的速率常数. 结果表明, 正丙苯高温氧化主要发生在烷基侧链, 包括6种C—C和C—H键断裂单分子分解反应以及3种侧链氢原子与氧气或其它小自由基的氢提取反应, 其中与苄基相连的C—C键具有最小断键能, 是最重要的单分子分解反应, 而不同位点的自由基氢提取反应的贡献相似; 体系的模拟温度和密度/压力与正丙苯的氧化速率呈正相关, 但当量比对氧化速率的影响则严重依赖于体系温度. 计算所得正丙苯高温氧化表观活化能和指前因子与文献报道的实验值相比在可接受的范围内.
正丙苯; 反应机理; 高温氧化; 反应力场; 分子动力学模拟
航空煤油作为航空发动机燃烧的主要能源, 如何提高其燃烧效率、 降低污染物的生成排放越发受到关注. 由于航空煤油包含多种烷烃、 烯烃、 环烷烃和芳香烃等碳氢组分, 很难直接构建其燃烧动力学模型. 通常采用能够表征航空煤油重要理化特性的替代燃料模型研究航空煤油的燃烧特性[1]. 在航空煤油的替代燃料模型研究中, 正丙苯作为芳香烃组分的典型替代燃料, 它具有足够长的烷基侧链(正丙基,-C3H7)和苯基(—C6H5), 可以研究侧链脂肪族性质和正丙苯的芳香族性质. 如, Dagaut等[2]构建的航空煤油替代燃料模型中正丙苯占26%, 徐佳琪等[3]开发的RP-3三组分替代燃料模型中正丙苯占15.8%, 而Dooley等[4]开发的Jet A燃料和第二代替代燃料模型中正丙苯占22.8%(摩尔分数). 此外, 与其它单环芳烃(如甲苯、 乙基苯和二甲苯)相比, 正丙苯氧化可以产生更多的小分子烃类(如甲烷、 乙烯和丙烯), 与丁苯及其它更大的烷基单取代苯相比, 其侧链较短能够有效减少动力学模型的构建规模. 因此, 为了深入理解正丙苯的燃烧特性, 研究人员对正丙苯展开了系列的实验、 动力学模拟和理论计算研究.
在实验方面, 研究主要关注正丙苯的层流燃烧速率[5~9]和点火延迟时间[10~13]. 这两类实验数据能够直观反映正丙苯燃烧过程的链诱发和链传递动力学特征, 对正丙苯燃烧动力学模型的开发和验证至关重要. 此外, 正丙苯燃烧热解过程的主要物种浓度分布为正丙苯燃烧动力学模型中关键反应网络及热动力学参数的合理性验证提供了更为丰富的实验依据[14~17]. 在动力学模拟方面, 对于正丙苯的动力学模型构建主要根据反应类规则, 以甲苯及乙苯的动力学模型为基础, 建立正丙苯的燃烧动力学模型[17,18], 在此基础上添加包括烷基过氧自由基和氢过氧烷基自由基的相关反应, 建立正丙苯的低温氧化动力学模型[12,19], 此外, 通过删除正丙苯燃烧模型中的含氧物种及相关反应, 建立正丙苯的热解动力学模型[14,15]. 在理论计算方面, 主要采取量子化学计算, 针对正丙苯燃烧裂解过程中最关键的初始消耗反应[即小自由基(H/CH3/HO2/O2)]与正丙苯的提取反应动力学开展研究[14,20~22].
随着先进发动机工况向极端条件(如超高温、 超高压)发展, 受限于反应器本身材料的性质、 使用环境的特殊性以及燃烧反应的快速性, 导致现有的燃烧实验诊断技术难以直接监测燃料反应过程中多个特定产物的时间演变(fs级). 虽然前述实验提供了正丙苯氧化的初始产物和有关反应的必要信息, 然而, 特别是在高温下, 氧化反应进展非常快, 并产生大量的中间体和活性自由基, 难以通过实验捕捉. 此外, 由于反应的时间尺度和空间尺度都很小, 从检测到的有限物质中推断出详细的氧化反应过程也不够全面. 因此, 采用理论方法研究极端条件下的燃料燃烧反应特性是一种较好的手段. 原则上, 量子化学计算可以提供准确的反应物、 过渡态和产物的结构信息, 但主要针对小分子燃料体系[23]. 对于复杂体系特别是大分子燃料体系, 量子化学计算所需的时间成本极其昂贵而受到限制.
由van Duin等[24]基于键级和键能关系开发的ReaxFF反应力场, 其力场参数完全来自对力场目标体系的量子化学计算结果的拟合, 可以快速准确地观察到化学过程中反应物、 中间体和产物的时间演化趋势, 为研究较大反应体系的物理化学性质提供了有效途径. 并且经过多年的发展, ReaxFF反应力场已被广泛应用于含能材料、 催化、 燃烧和聚合物等多个研究领域[25]. 为了更加准确地描述碳氢燃料的燃烧过程, Chenoweth等[26]基于量子化学计算结果优化了ReaxFF反应力场的力场参数, 并对一系列的碳氢燃料燃烧特性进行模拟, 结果表明, ReaxFF反应力场可以正确预测碳氢燃料的反应活性趋势[27]. 此外, 其他一些研究者也使用该力场参数研究了正十二烷、 甲苯、 RP-3和乙烯的燃烧特性, 其模拟结果与密度泛函理论方法计算结果和可测量的实验结果也相吻合[28~31]. 综上表明, ReaxFF反应力场适合于碳氢燃料的燃烧特性研究.
本文采用ReaxFF反应力场对正丙苯高温氧化的起始反应路径、 主要产物的形成机理以及不同温度、 密度和当量比下正丙苯高温氧化的主要产物分布规律进行了分子动力学模拟研究, 并采用反应动力学方法计算了正丙苯的高温氧化动力学参数.
1 计算方法与模拟细节
1.1 ReaxFF反应力场
ReaxFF反应力场是由van Duin等[24]基于第一性原理提出的, 以原子间的键级为核心, 通过键级、 键长和键能之间的关系以及原子间电荷的动态传递来模拟原子之间化学键的生成和断裂. 在分子动力学的计算过程中, 当化学键断裂时, 与价键相关的力和能量变为零, 因此该反应力场能够描述化学键的生成和断裂. ReaxFF反应力场的计算表达式如下:
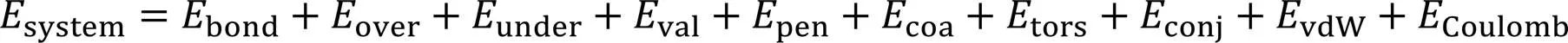
上式主要由两大部分组成: 基于键级的成键相互作用能量项(kJ/mol): 键能项(bond)、 配位修正项 (over和under)、 键角能项(val)、 惩罚能项(pen)、 三体共轭项(coa)、 二面角共轭作用能项(tors)和四面体共轭项(conj)等; 非键相互作用能项(kJ/mol): 范德华相互作用能项(vdW)和库仑力相互作用能项(Coulomb).
初始分子结构采用密度泛函理论(DFT)方法B3LYP泛函结合6-311++G(,)基组进行优化. 在ReaxFF模拟中, 在5 K时进行能量最小化, 时间步长为0.1 fs, 原子数()、 体积()和温度()恒定, 表示为NVT-MD(分子动力学模拟). 然后使用NVT-MD模拟, 将这些系统在5 K下驰豫, 使得模拟体系达到平衡, 平衡50 ps, 时间步长为0.1 fs. 时间步长的设置主要参考Chenoweth等[26]的工作. 在所有 NVT-MD模拟过程中, 采用Berendsen方法[32]对温度和压力进行调节, 耦合参数为500 fs, 使温度在设定值附近波动. 将键级设定为0.3, 当键级大于0.3时, 判定新化学键形成, 产物生成. 当量比()由正丙苯实际氧化过程的正丙苯/氧气的摩尔比与正丙苯完全燃烧所需正丙苯/氧气的摩尔比之比确定, 体系密度通过控制立方体体积设置, 所有输入条件的汇总见表1, 其范围从在最低温度和密度(= 2300 K,=0.05 g/cm3,=2.0)到最高温度和密度(=3500 K,=0.35 g/cm3,=0.5), 关于体系密度设置主要参考已报道的类似反应体系[27,28,33], 本文通过理想气体状态方程, 计算出的压力范围为22~286 MPa. 注意提高模拟温度研究燃料燃烧和热解是ReaxFF模拟的常见做法, 旨在通过提高温度来缩短反应时间, 同时不改变反应路径, 又保留原本温度下的动力学行为[34]. 本文采用的是Chenoweth等[26]报道的CHO-2008力场参数. 目前, 该研究小组有更新改进的CHO-2016力场参数[35], 该力场参数与 CHO-2008相比, 保持了CHO-2008对大分子碳氢化合物的模拟精度, 主要改善了CHO-2008对C1化学的预测精度. 值得注意的是, 文献[35]进一步验证了使用CHO-2016与CHO-2008研究正丁基苯热解初始反应机理, 表明CHO-2016与CHO-2008模拟结果一致. 最近, Lindgren等[33]采用CHO-2008探究环戊烷和环戊烯的燃烧初始反应机理, 他们也指出CHO-2016主要改善C1化学, 对于研究较大的碳氢化合物, CHO-2008也是可以的. 因此, 选择CHO-2008研究正丙苯的高温氧化机理. 此外, 采用Packmol程序[36]构建周期性体系, 并且所有的MD模拟都在LAMMPS软件[37]中完成, 使用ChemTrayzer程序[38]进行反应路径的后处理.
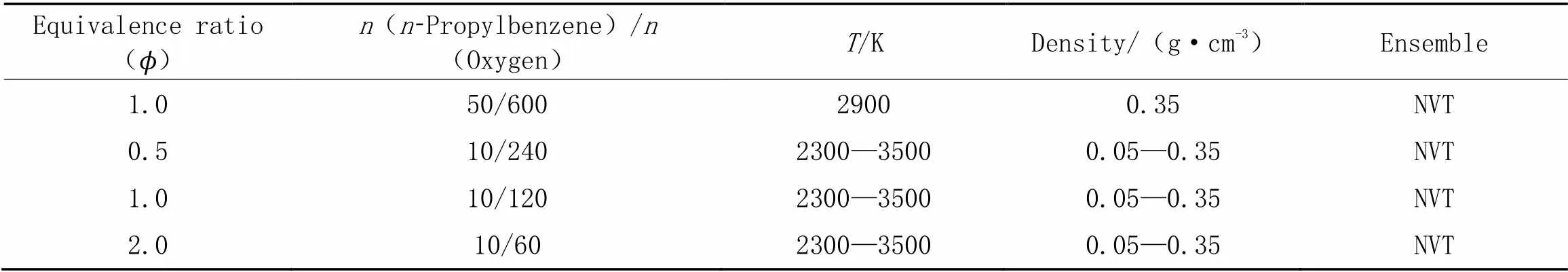
Table 1 Temperature, density, and equivalence ratio of n-propylbenzene oxidation system
1.2 DFT计算
为了验证反应力场的合理性和可迁移性, 同时采用DFT计算和ReaxFF模拟研究了正丙苯氧化过程中侧链C—C键的键解离能, 为了分析正丙苯燃烧化学动力学过程中的初始氢提取反应, 采用DFT 计算了正丙苯不同碳原子位点上的C—H键的键能. 所有分子和自由基的几何结构优化采用DFT 方法B3LYP泛函结合6-311++G(,)基组进行. DFT计算均采用Guassian 16量子化学计算软件包[39]完成.
2 结果与讨论
2.1 正丙苯氧化的初始反应机理
为了确定正丙苯氧化的初始反应机理, 尽量减小由于反应体系过小、 反应物的数量过少带来结果的差异过小而不能显著观察出初始反应通道, 将50个正丙苯分子和600个氧气分子放置在温度为2900 K、 密度为0.35 g/cm3条件下进行NVT⁃MD模拟. 通过分析原子成键情况以及在不同时刻下的氧化产物, 最终得到了正丙苯高温氧化的初始反应途径. 正丙苯初始反应主要可分为两大类: 正丙苯侧链上发生裂解反应以及与其它自由基发生的提取反应. 图1展示了观察到的正丙苯发生裂解的4条反应路径及频率. 正丙苯链上发生C—C断裂生成苄基和乙基(CH3CH2)是最主要的裂解反应, 反应频率为25次, 占到正丙苯初始消耗的一半. 其次是C—C断裂生成C6H5CH2CH2和CH3自由基, 反应频率为 5次. 裂解反应中除C—C键的断裂外, C—H键的断裂也比较重要, 正丙苯通过断裂侧链上不同碳位上的C—H键生成不同的自由基: C6H5CH2CH2CH2自由基和C6H5CH2CHCH3自由基, 频率分别为2和1. 如 图2所示, 正丙苯与不同自由基及氧气分子发生反应, 模拟中共发现了9条不同的反应路径, 总频率为17次. 值得注意的是, 氢提取反应频率就占到了14次. 上述结果表明正丙苯消耗的初始反应主要为裂解反应与氢提取反应. 由于裂解反应在正丙苯初始消耗中至关重要. 因此, 使用DFT方法和ReaxFF方法计算了正丙苯侧链上不同C原子上C—H和C—C键的键能.
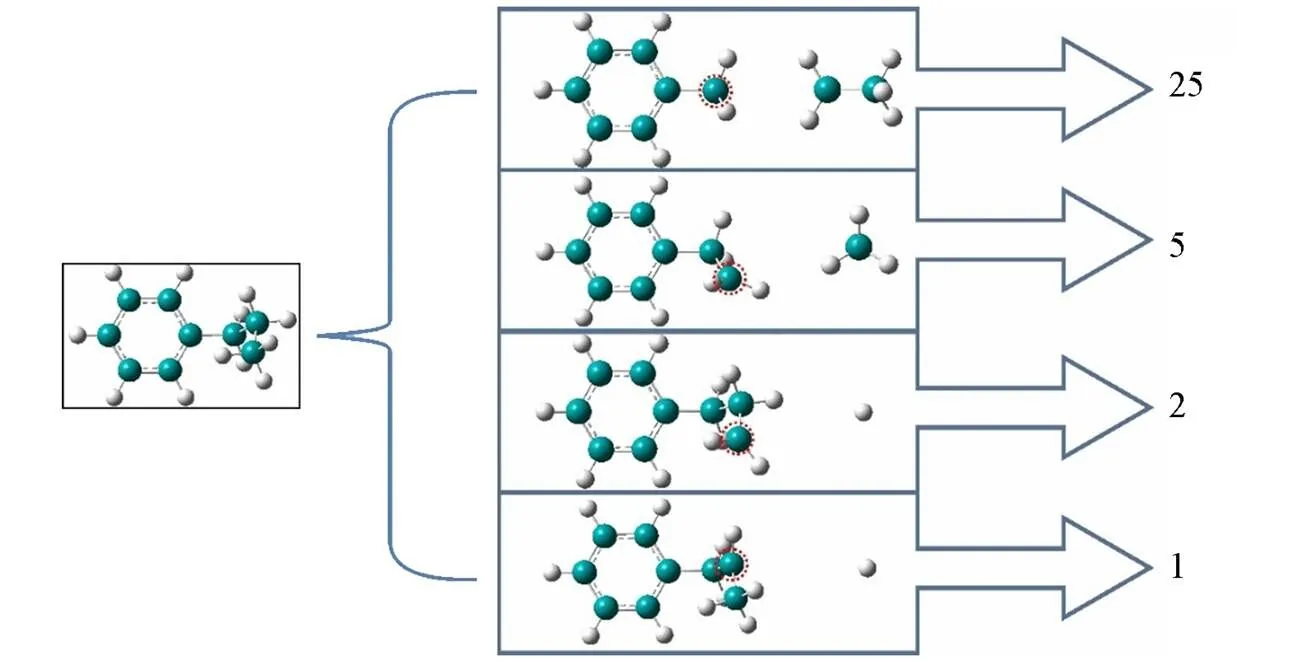
Fig.1 Four reaction pathways of n⁃propylbenzene pyrolysis
Green and white spheres represent carbon and hydrogen atoms, respectively.
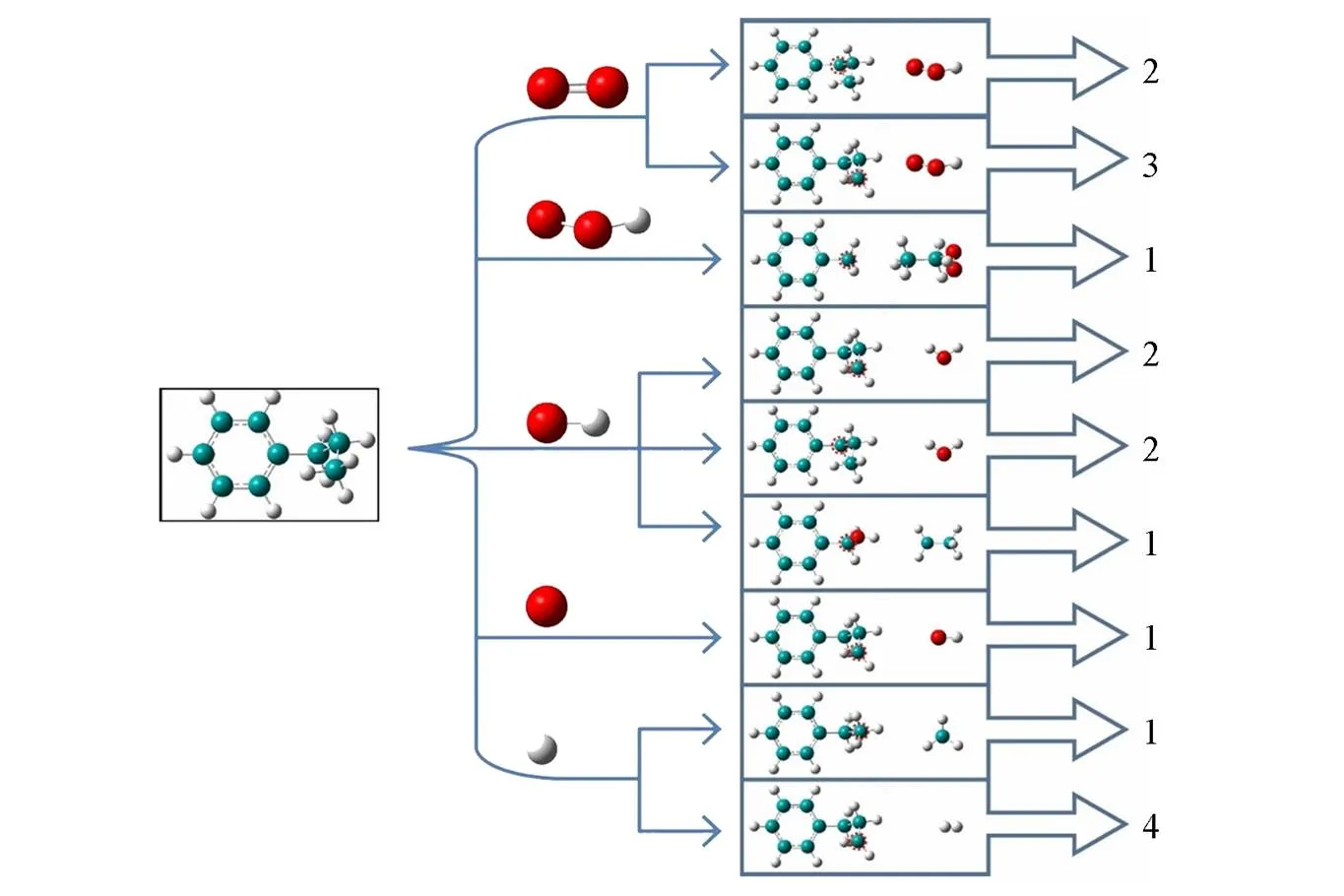
Fig.2 Nine reaction pathways of n⁃propylbenzene reacting with different small radicals and oxygen
Green, red and white spheres represent carbon, oxygen and hydrogen atoms, respectively.
结果如表2[17,40]所示, 与苄基相连的C—C键的键能最小. 因此在反应过程中与苄基相连的C—C键最易断裂, 这与上述模拟结果相符. 碳氢化合物燃烧的第一步是通过打破单个C—C键来分解燃料分子[41], C—C键通常比C—H弱[40]. 此外, 可以看出, 不论是B3LYP还是ReaxFF, 与Wang等[17]采用高精度CBS-QB3方法计算的正丙苯单分子分解主要断键能和Luo等[40]的断键能数据库相比, 结果均吻合较好, 说明采用ReaxFF研究正丙苯高温氧化反应体系是合适的.
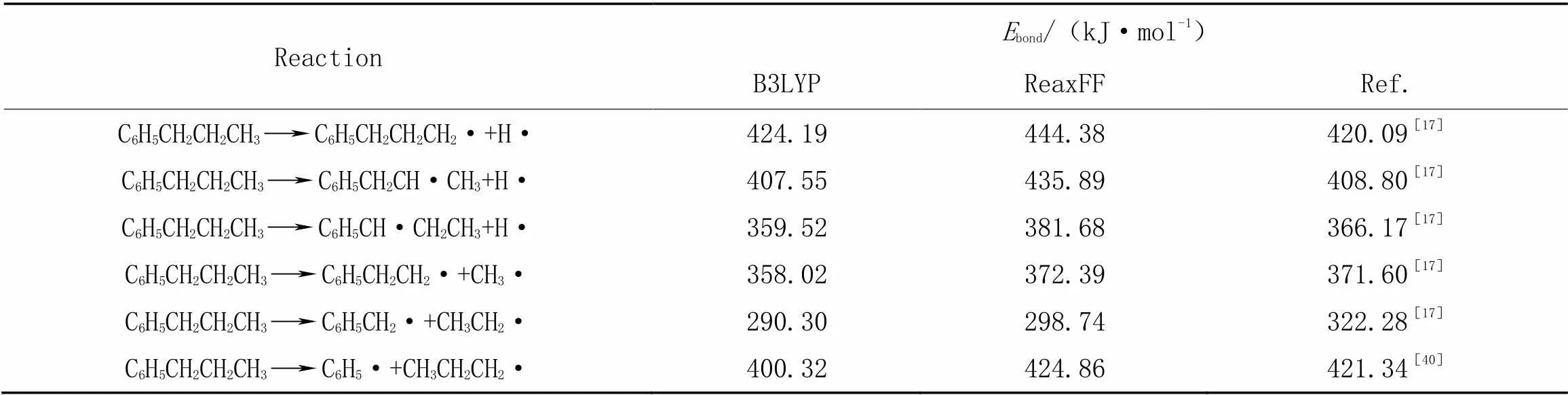
Table 2 Calculated bond dissociation energies of n-propylbenzene using B3LYP and ReaxFF methods and the corresponding literature results
通过ChemTraYzer程序[38]对生成的键级信息进行后处理, 获得特定目标组分的每一步反应, 再将反应物和生成物串联即得到正丙苯的主要反应网络, 如Scheme 1所示, 它提供了从最初反应到最终产物反应机理的详细描述. 结果显示, 无论是正丙苯(PBZ)直接发生裂解还是与其它自由基反应形成苯丙基自由基(PBZJA), 随后大部分都会生成苄基(PHCH2), 这与实验结果一致[42], 说明PHCH2是非常重要的中间体. 之后PHCH2与HO2自由基结合生成C7H8O2分子(PHCH3O2), 然后O—O键迅速断裂生成苯甲醇自由基(PHCH2O)和OH自由基, 或直接与O自由基结合生成PHCH2O. PHCH2O的C—C键断裂生成苯基(PHJA)和甲醛, PHJA氧化生成C6H5O2自由基(PHO2), 并且进一步氧化生成C6H5O自由基(PHO). 接下来PHO异构化形成五元环自由基(CPDCO), 这是苯环消耗的重要反应之一[1]. 之后裂解形成环戊二烯自由基(CPDJA)和CO. 最后, CPDJA发生开环反应, 并且进一步氧化生成CO, CO2和H2O.
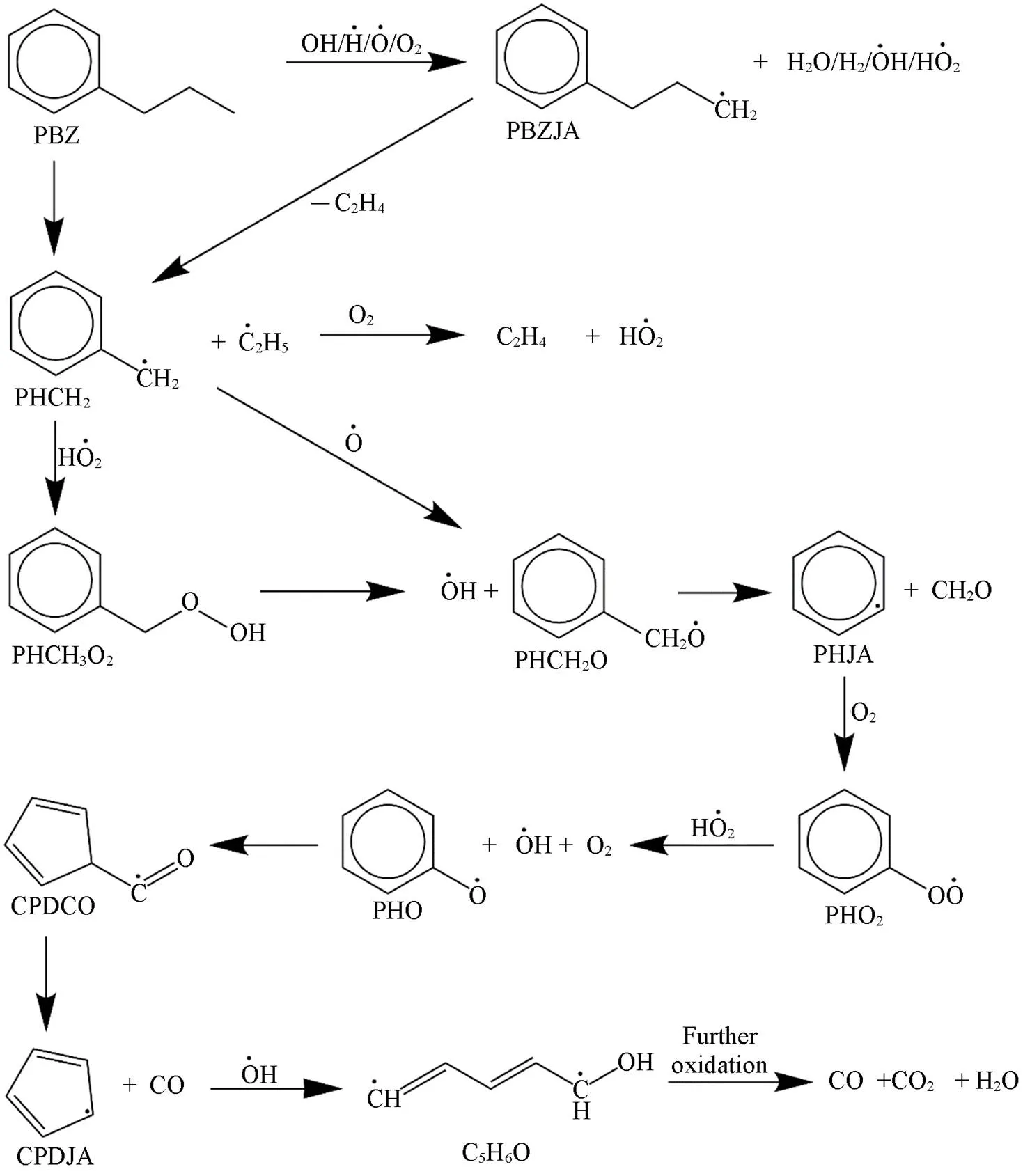
Scheme 1⁃Propylbenzene partial oxidation pathway at 2900 K
2.2 主要氧化产物的形成机理
尽管已经对正丙苯的氧化进行了广泛研究, 但对中间机制的直接描述仍然十分困难. 由于对主要产物相关反应的详细分析对于理解中间机理尤其重要, 因此, 对正丙苯氧化过程中H2O, CH2O, CO和CO2的反应途径进行了分析. 反应的体系为3500 K, 0.35 g/cm3,=2.0,=330. 图3介绍了正丙苯氧化过程中H2O, CH2O, CO和CO2的反应途径. 在图3(A)中, H2O主要是通过体系中的H和OH自由基与其它分子反应形成, 最主要的反应通道是H2+OH→H2O+H. 此外, 含氧自由基直接裂解生成H2O也是重要的反应通道(如C2H3O→H2O+C2HO). 在模拟过程中,H2O主要是通过被其它分子提取H或OH而被消耗, 或H2O的直接分解(H2O→H+OH).
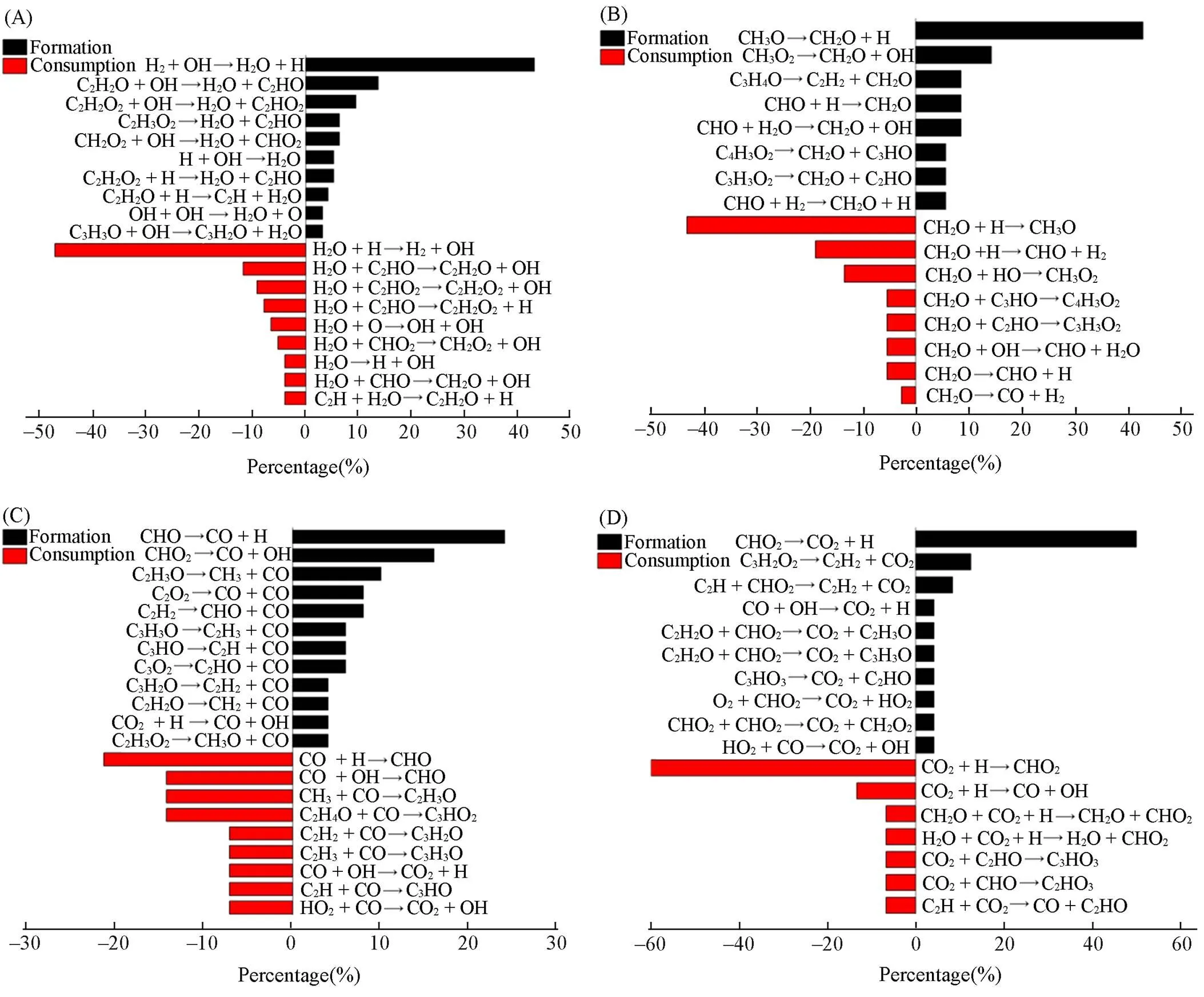
Fig.3 Formation and consumption reaction pathways of the main oxidation products H2O(A), CH2O(B), CO(C) and CO2(D)
对于碳氢化合物的化学动力学模拟研究, 甲醛(CH2O)被证实为碳氢化合物高温燃烧的重要中间组分, 直接影响到最后产物CO2和CO等的形成[28]. 在图3(B)中, CH2O主要是由甲氧基(CH3O)和过氧甲基(CH3O2)的裂解形成(CH3O→CH2O+H, CH3O2→CH2O+OH), 而CH2O主要是与H自由基反应被消耗(CH2O+H→CH3O, CH2O+H→CHO+H2).
对于碳氢化合物的氧化, CO和CO2是主要的含碳稳定产物, 该反应在整个氧化机理中起着重要作用. 由图3(C)可见, CO的形成主要是通过体系中的含氧自由基裂解形成(如CHO→CO+H, CHO2→CO+HO, C2H3O→CH3+CO). CO的消耗主要基于CO与其它自由基复合形成含氧自由基 (如CO+H→CHO, CO+HO→CHO). 由图3(D)可见, 相比于CO, CO2的形成与CHO2更加密切, 主要反应方式为CHO2直接发生裂解(CHO2→CO2+H)以及CHO2与各类自由基及分子发生氢提取反应形成(如C2H+CHO2→C2H2+CO2, C2H2O+CHO2→CO2+C2H3O), CO2主要是与H自由基反应而被消耗(CO2+H→CHO2, CO2+H→CO+HO).
2.3 温度对正丙苯氧化的影响
为了分析温度对正丙苯氧化的影响, 在不同条件(2300~3500 K、 间隔300 K,=0.35 g/cm3,=2.0,=330)下进行了一系列NVT-MD模拟, 模拟时长1 ns. 总分子数、 正丙苯分子数、 主要产物以及势能随时间的变化如图4所示. 可见, 势能衰减率随温度的升高下降得更快[图4(A)], 表明温度加速了正丙苯的氧化. 这种现象的原因可以用系统的热释放率来解释, 取决于温度[25]. 在模拟初始阶段, 尤其是在高温条件下, 势能会先升高然后迅速降低, 结合总分子数和正丙苯分子数变化情况 [图4(B)和(C)], 可以确定整个系统开始迅速吸收热量发生初始热解反应. 并且势能达到最大值的时间随温度的降低而增加, 表明热解反应需要更多时间. 总分子数的变化也表明, 随着模拟温度的升高, 反应进行得更快, 正丙苯分子的转化率也迅速增加. 从模拟结果来看, 主要的初始反应是正丙苯的热解和氢提取反应. 最初的反应产物和副产物随后与氧气反应, 释放出大量的能量, 最终导致势能急剧下降. 模拟结果显示, 最终的主要产物为CO, CO2和H2O. 并且随温度的升高, 它们出现得更早, 这表明温度升高导致产物形成时间提前. 产物分布表明, H2O和CO的含量随温度的升高而增加, 在3500 K时达到最大值[图4(D)~(F)]. 可见, 在当量比为0.5和1.0时, CO2与H2O和CO的分布具有相同趋势, 但在当量比2.0时, CO2的分布情况在高温下却表现出先增加后降低的趋势, 这是因为随着氧化的进行, CO2与系统中的自由基反应转化为CO和H2O, 这可能是因为与当量比为0.5和1.0时的模拟相比, 氧分子的浓度较低. 当温度在2300~2900 K范围内时, 主要产物的最终数量急剧增加, 但随着温度的持续升高, 主要产物的数量没有显著变化. 这表明当温度达到较高范围时, 温度升高对氧化过程的影响有限, 这一点从势能、 总分子数和正丙苯分子数的变化情况上同样得到验证.
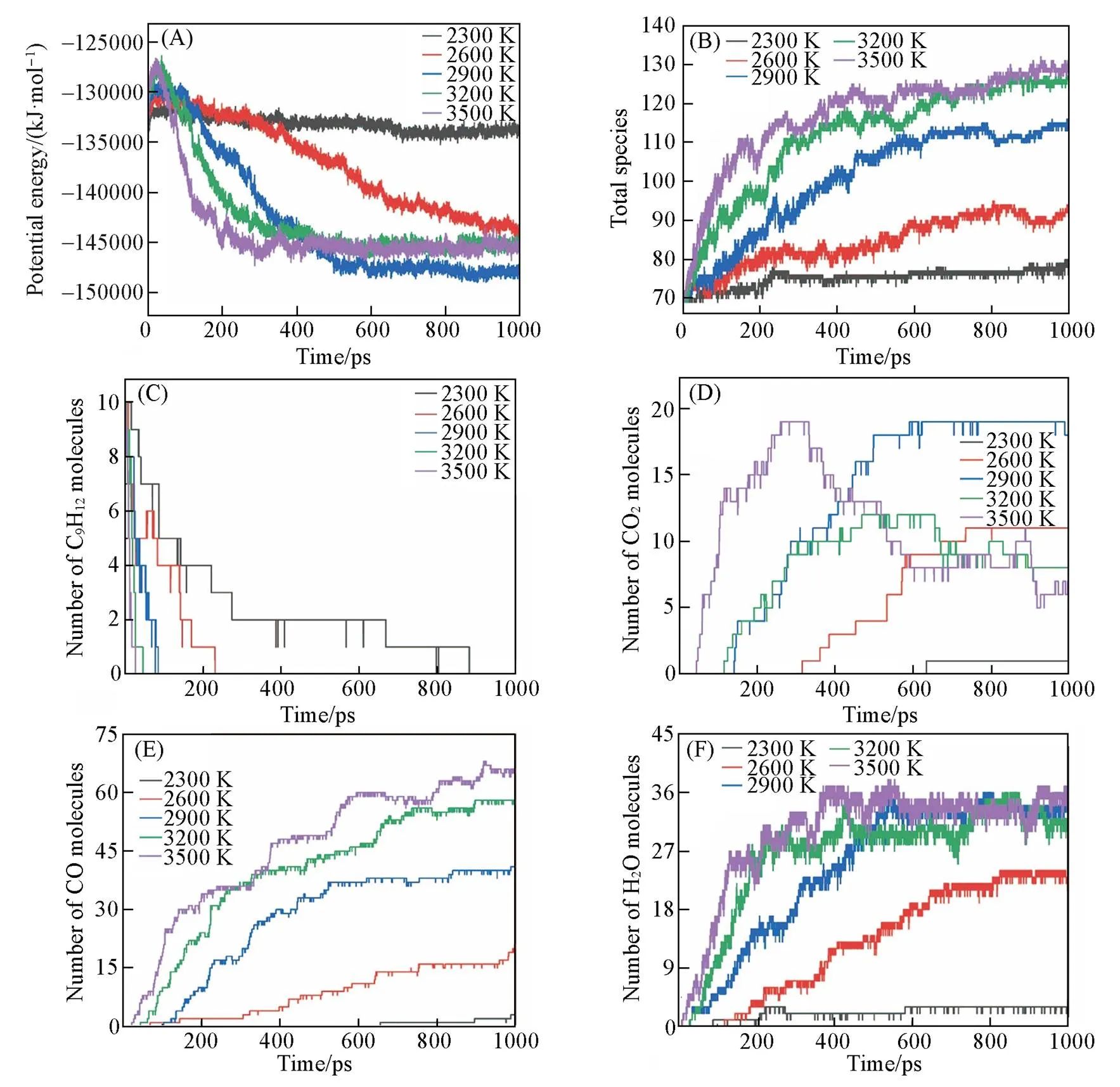
Fig.4 Time evolution of the potential energies(A), total number of species(B), n⁃propylbenzene(C) and main products of CO2(D), CO(E), H2O(F) at different temperatures(ρ=0.35 g/cm3, ϕ=2.0)
2.4 密度/压力对正丙苯氧化的影响
为了探究密度/压力对正丙苯氧化的影响, 分析了3个不同密度的模拟体系在温度为3500 K时的结果(=330,=2.0). 图5给出了在3500 K模拟条件下, 势能、 总分子数、 正丙苯分子数及主要产物随时间的变化. 由图5(A)可见, 随着模拟体系中密度的增加, 势能下降的趋势也增加, 并且密度为0.15和0.35 g/cm3的模拟体系能在1 ns达到热力学平衡. 图5(B)和(C)分别为总分子数和正丙苯分子数变化的趋势, 密度的增加导致反应的提前开始和燃料的快速消耗. 最终产物的分布也可以反映不同密度的影响. 由图5(D)和(E)可见, 产物H2O和CO随体系密度的增加而增加, 在0.35 g/cm3时达到最大值. 由图5(F)可见, CO2首先在0.35 g/cm3出现, 其最大分子数出现在0.15 g/cm3, 并且在不同密度下CO2的分子数始终低于CO, 与Yuan等[43]的实验结果一致. 这主要是由于体系中氧分子含量不足, 难以支撑大量CO转化为CO2. 通过整体观察得知, 密度/压力对于模拟结果的影响不如温度大.
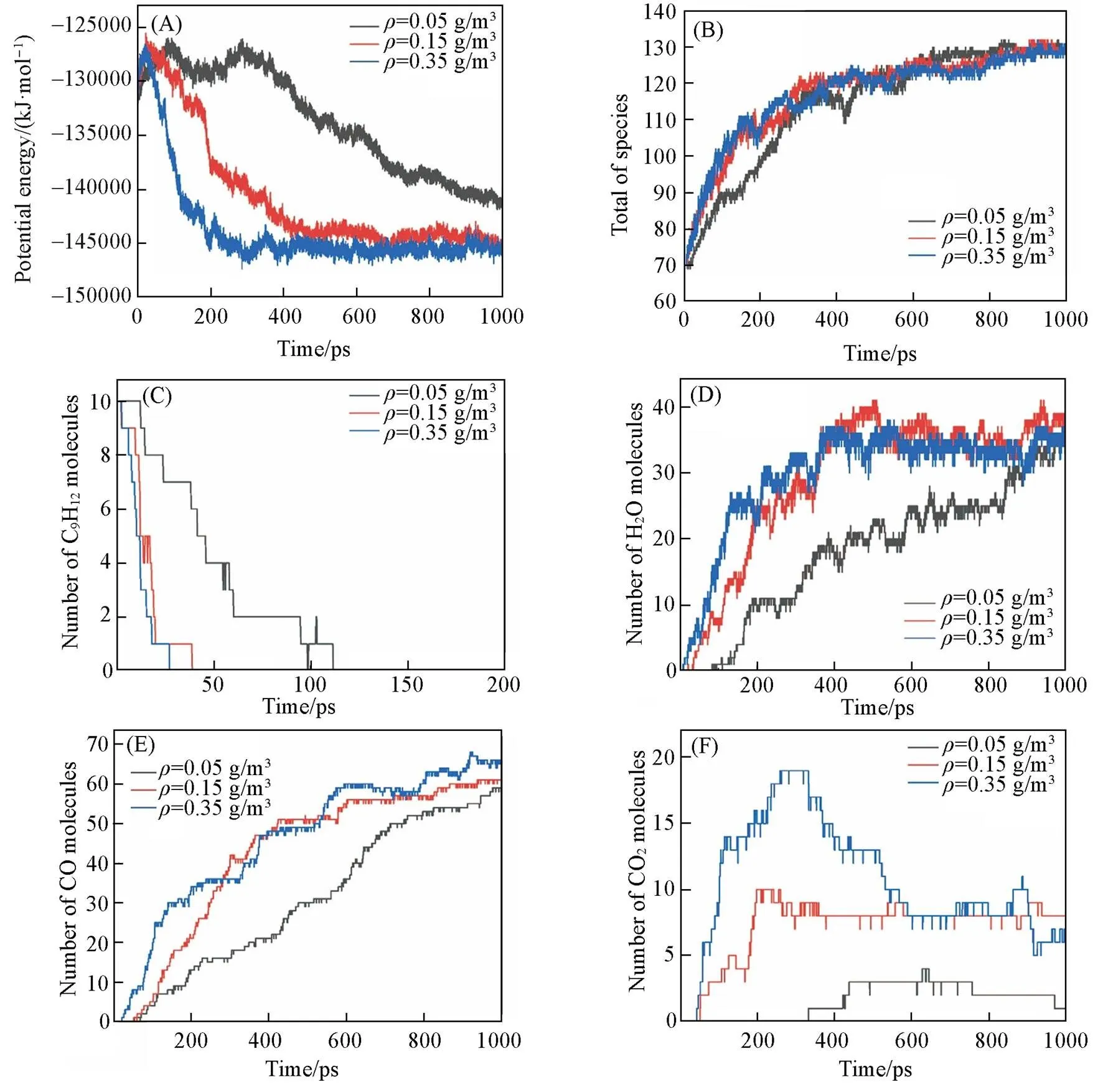
Fig.5 Time evolution of the potential energies(A), total number of species(B), n⁃propylbenzene(C), and main products of H2O(D), CO(E), CO2(F) at different densities(T=3500 K, ϕ=2.0)
2.5 当量比对正丙苯氧化的影响
为了分析不同当量比对正丙苯氧化的影响, 在3种不同当量比(=690,=0.5;=450,=1.0;=330,=2.0)下进行了一系列NVT-MD模拟. 并且是在3种不同密度(0.05, 0.15和0.35 g/cm3)、 温度范围2300~3500 K(间隔300 K)下进行. 在每次模拟中, 确定了燃料初始消耗所需的时间以及燃料分子消耗一半所需的时间. 模拟结果如图6(A)~(C)所示, 在较低的温度和密度下, 燃料初始消耗以及燃料分子消耗一半所需的时间较长. 表明正丙苯在该模拟工况下反应活性较小. 上述已经讨论了温度和密度对反应的影响, 因此, 将进一步关注当量比对正丙苯燃烧反应的影响. 显然当量比对结果有一定的影响, 但其影响作用根据模拟温度不同会产生较大变化,在高温条件下(≥2900 K), 结果基本没有差别, 而在低温条件下, 模拟结果产生较大差异, 与Cheng等[29]模拟的结果相类似. 图7为在不同当量比下模拟得到的主要产物随时间的变化趋势(NVT-MD模拟,=3500 K,=0.35 g/cm3), 由图7(A)和(B)可见, 随着当量比的降低, 产物CO2的含量分布逐步增加, CO的分子数呈现一个先增加后降低的趋势(≥1.0). 与此同时, CO2增加. 由图7(C)可见, 当=0.5和1.0时, 产物H2O的含量基本一致, 而当=2时, H2O的含量却相对较低, 这主要是氧气含量不足所致.
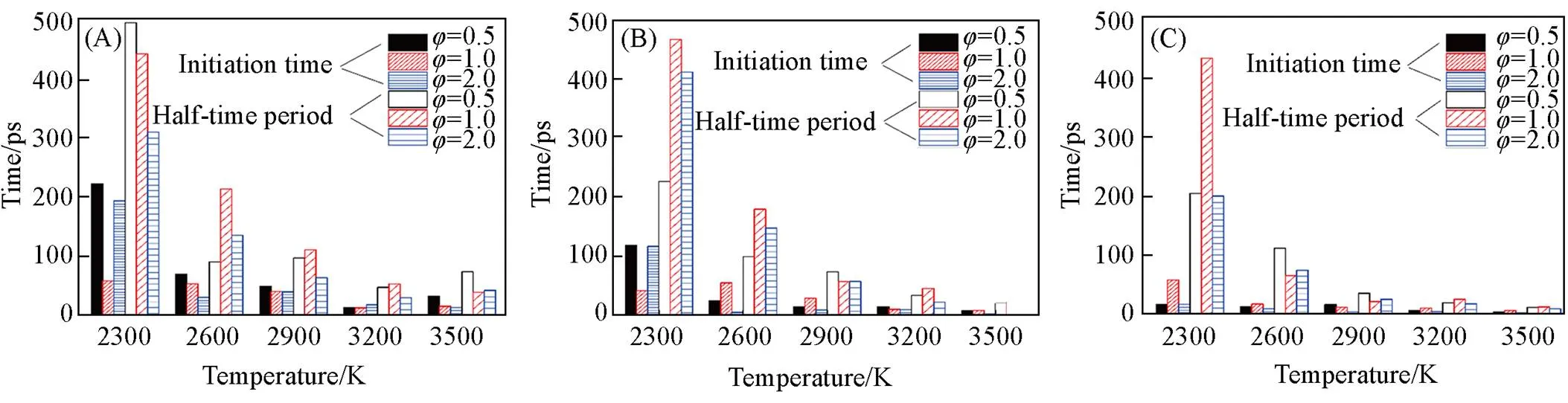
Fig.6 Temperature evolution of the initiation time as well as half⁃time period of n⁃propylbenzene oxidation at different densities of 0.05(A), 0.15(B), 0.35 g/cm3(C) and equivalence ratios
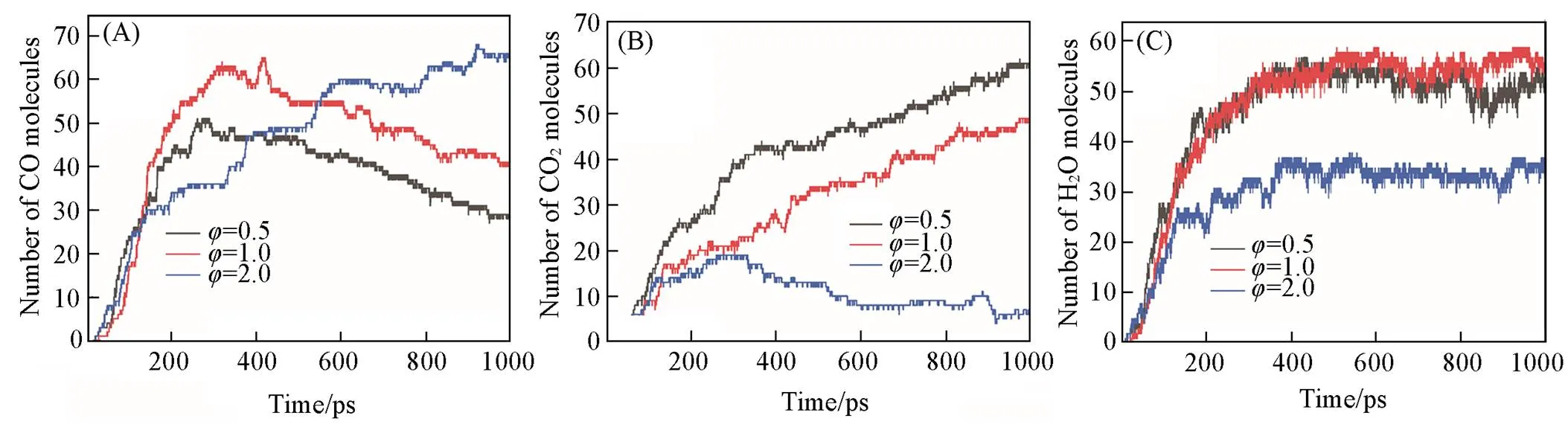
Fig.7 Time evolution of the main products of CO(A), CO2(B), H2O(C) of n⁃propylbenzene oxidation at different equivalence ratios(T=3500 K, ρ=0.35 g/cm3)
2.6 正丙苯氧化的动力学
正丙苯完全氧化反应(C9H12+12O2=9CO2+6H2O), 在2300~3500 K(间隔300 K)温度范围内、 3种不同密度(0.05, 0.15和0.35 g/cm3)、 当量比为0.5~2.0(=330, 450和690)下对正丙苯/氧气系统进行NVT-MD模拟. 每个温度下反应的总速率常数(, cm3·mol-1·s-1)可以通过式(2)来简化, 式(2)为正丙苯氧化的二级反应动力学微分方程, 主要假设是将反应物正丙苯和氧气均作为一级反应处理, 即
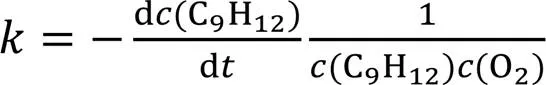
进一步采用不同温度下拟合得到的速率常数通过Arrhenius方程式获得正丙苯氧化的总反应的表观活化能(a)和指前因子()等Arrhenius参数, 具体数值列于表3.
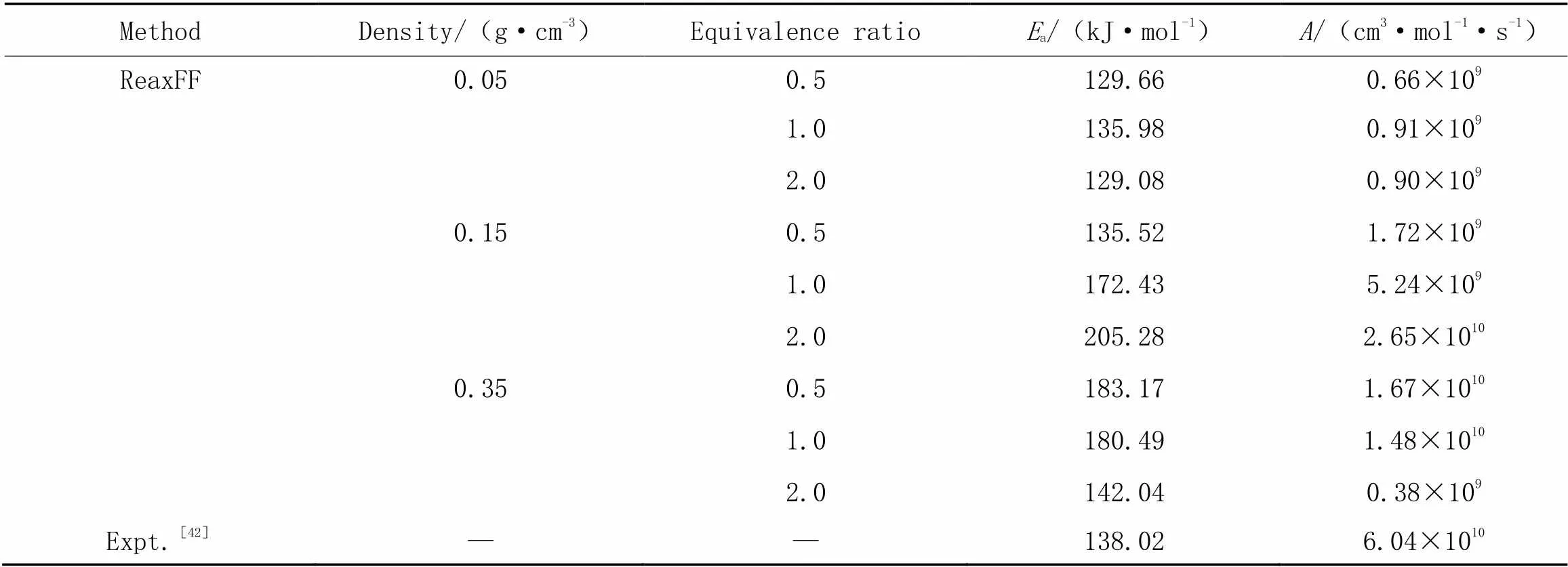
Table 3 Fitted Arrhenius parameters including Ea and A
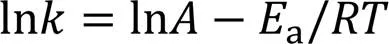
从表3可见, 拟合得到的Arrhenius参数与Liang等[42]的实验结果相比也在可以接受范围内, 表明基于ReaxFF反应力场的分子动力学模拟的合理性.
3 结 论
采用DFT计算和ReaxFF模拟相结合的方法对正丙苯高温氧化机理进行研究, 并对初始反应进行了分析. 正丙苯的初始反应主要通过两种途径: (1) 正丙苯侧链上C—C和C—H键的断裂; (2) 正丙苯与氧气分子或其它自由基发生氢提取反应, 其中, 侧链中与苄基相连的C—C键的键能最小, 因此在反应过程中最易断裂, 是主要的起始反应路径. 依据模拟结果, 给出了正丙苯高温氧化过程的主要反应网络, 并且发现无论是通过哪一种初始反应, 随后大部分都会生成苄基, 说明苄基是非常重要的中间体. 在主要产物CO, CO2和H2O的形成机理分析中, H2O与体系中H和OH自由基紧密相关, CH2O主要是由CH3O和CH3O2的裂解形成,直接影响到最后产物CO和CO2等的形成,而主要的含碳稳定产物CO和CO2的生成和消耗主要由体系中CHO2自由基决定. 增加体系的密度/压力和模拟温度可以很大程度上加快正丙苯的氧化速率、 加速产物的形成. 但当温度达到较高范围时, 温度升高对氧化过程的影响有限. 相对而言, 密度/压力对结果的影响要小于温度. 与温度和密度/压力的影响不同, ReaxFF模拟结果表明, 在低温下当量比对正丙苯消耗的反应速率的影响较大, 而高温条件下对模拟结果影响较小. 对正丙苯高温氧化进行了反应动力学分析, 计算得到的表观活化能和指前因子与实验结果相比也在可接受的范围内.
[1] Liu Y. X., Wang B. Y., Weng J. J., Yu D., Richter S., Kick T., Tian Z. Y.,, 2018,, 53—65
[2] Dagaut P., El Bakali A., Ristori A.,, 2006,(7/8), 944—956
[3] Xu J. Q., Guo J. J., Liu A. K., Wang J. L., Tan N. X., Li X. Y.,, 2015,(4), 643—652(徐佳琪, 郭俊江, 刘爱科, 王健礼, 谈宁馨, 李象远. 物理化学学报, 2015,(4), 643—652)
[4] Dooley S., Won S. H., Heyne J., Farouk T. I., Ju Y., Dryer F. L., Brezinsky K.,, 2012,(4), 1444—1466
[5] Johnston R. J., Farrell J. T.,, 2005,(1), 217—224
[6] Hui X., Das A. K., Kumar K., Sung C. J., Dooley S., Dryer F. L.,, 2012,, 695—702
[7] Hui X., Sung C. J.,, 2013,, 191—200
[8] Ji C., Dames E., Wang H., Egolfopoulos F. N.,, 2012,(3), 1070—1081
[9] Mehl M., Herbinet O., Dirrenberger P., Bounaceur R., Glaude P. A., Battin⁃Leclerc F., Pitz W. J.,, 2015,(1), 341—348
[10] Roubaud A., Minetti R., Sochet L. R..,, 2000,(3), 535—541
[11] Darcy D., Mehl M., Simmie J. M., Würmel J., Metcalfe W. K., Westbrook C. K., Curran H. J.,, 2013,(1), 411—418
[12] Darcy D., Nakamura H., Tobin C. J., Mehl M., Metcalfe W. K., Pitz W. J., Curran H. J.,, 2014,(1), 65—74
[13] Darcy D., Nakamura H., Tobin C. J., Mehl M., Metcalfe W. K., Pitz W. J., Curran H. J.,, 2014,(6), 1460—1473
[14] Gudiyella S., Brezinsky K.,, 2012,(3), 940—958
[15] Gudiyella S., Brezinsky K.,, 2013,(1), 1767—1774
[16] Anderson H., McEnally C. S., Pfefferle L. D.,, 2000,(2), 2577—2583
[17] Wang Z., Li Y., Zhang F., Zhang L., Yuan W., Wang Y., Qi F.,, 2013,(1), 1785—1793
[18] Li Y., Cai J., Zhang L., Yang J., Wang Z., Qi F.,, 2011,(1), 617—624
[19] Darcy D., Tobin C. J., Yasunaga K., Simmie J. M., Würmel J., Metcalfe W. K., Tidjani N., Ahmed S. S., Westbrook C. K., Curran H. J.,e, 2012,(7), 2219—2232
[20] Robinson R. K., Lindstedt R. P.,, 2013,(12), 2642—2653
[21] Altarawneh M., Dlugogorski B. Z.,, 2015,(4), 1406—1416
[22] Zhou C. W., Simmie J. M., Somers K. P., Goldsmith C. F., Curran H. J.,, 2017,(9), 1890—1899
[23] Shang Y., Ning H., Shi J., Luo S. N.,, 2021,(3), 711—717
[24] van Duin A. C. T., Dasgupta S., Lorant F., Goddard W. A.,, 2001,(41), 9396—9409
[25] Li X., Zheng M., Ren C., Guo L.,, 2021,(15), 11707—11739
[26] Chenoweth K., van Duin A. C. T., Goddard W. A.,, 2008,(5), 1040—1053
[27] Chenoweth K., van Duin A. C. T., Dasgupta S., Goddard W. A.,, 2009,(9), 1740—1746
[28] Wang Q. D., Wang J. B., Li J. Q., Tan N. X., Li X. Y.,, 2011,(2), 217—226
[29] Cheng X. M., Wang Q. D., Li J. Q., Wang J. B., Li X. Y.,, 2012,(40), 9811—9818
[30] Liu X. L., Li, X. X., Han S., Qiao X. J., Zhong B. J., Guo L.,, 2016,(6), 1424—1433(刘晓龙, 李晓霞, 韩嵩, 乔显杰, 钟北京, 郭力. 物理化学学报, 2016,(6), 1424—1433)
[31] Liu J. X., Min J., Xu H. J., Ren H. S., Tan N. X.,, 2022,(4),20210834(刘嘉欣, 闵杰, 许华杰, 任海生, 谭宁馨. 高等学校化学学报, 2022,(4),20210834)
[32] Berendsen H. J., Postma J., Gunsteren W., DiNola A. D., Haak J. R.,., 1984,(8), 3684—3690
[33] Lindgren E. B., Monteiro J. G. S., dos Santos A. R., Fleming F. P., Barbosa A. G. H.,, 2021,, 121205
[34] So M. R., Rensen., Voter A. F.,, 2000,(21), 9599—9606
[35] Ashraf C., van Duin A. C. T.,, 2017,(5), 1051—1068
[36] Martínez L., Andrade R., Birgin E. G., Martínez J. M.,, 2010,(13), 2157—2164
[37] Plimpton S.,, 1995,(1), 1—19
[38] Döntgen M., Przybylski⁃Freund M. D., Kröger L. C., Kopp W. A., Ismail A. E., Leonhard K.,, 2015,(6), 2517—2524
[39] Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Petersson G. A., Nakatsuji H., Li X., Caricato M., Marenich A. V., Bloino J., Janesko B. G., Gomperts R., Mennucci B., Hratchian H. P., Ortiz J. V., Izmaylov A. F., Sonnenberg J. L., Williams⁃Young D., Ding F., Lipparini F., Egidi F., Goings J., Peng B., Petrone A., Henderson T., Ranasinghe D., Zakrzewski V. G., Gao J., Rega N., Zheng G., Liang W., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Throssell K., Montgomery J. A. Jr., Peralta J. E., Ogliaro F., Bearpark M. J., Heyd J. J., Brothers E. N., Kudin K. N., Staroverov V. N., Keith T. A., Kobayashi R., Normand J., Raghavachari K., Rendell A. P., Burant J. C., Iyengar S. S., Tomasi J., Cossi M., Millam J. M., Klene M., Adamo C., Cammi R., Ochterski J. W., Martin R. L., Morokuma K., Farkas O., Foresman J. B., Fox D. J.,16.,. 01, Gaussian Inc., Wallingford CT, 2016
[40] Luo Y. R.,, CRC Press, London, 2007
[41] Kirkpatrick A. T.,, John Wiley & Sons, Hoboken, 2020
[42] Liang J., Li F., Cao S., Li X., Jia M. X., Wang Q. D.,, 2022,, 124940
[43] Yuan W., Li Y., Dagaut P., Wang Y., Wang Z., Qi F.,, 2017,, 178—192
Molecular Dynamics Simulation Study on High Temperature Oxidation Mechanism of-Propylbenzene
ZHOUZihao, WANGSihao, HUANGDaichuan, LIUBo, NINGHongbo*
(,,,610031,)
-Propylbenzene is a typical aromatic substitute component of Jet A, Jet A-1 and RP-3 aviation kerosene. In this work, the main oxidation reaction networks and the product distributions of-propylbenzene at different temperatures, densities and equivalence ratios were investigated by ReaxFF based on reactive molecular dynamics simulation. The reaction kinetics theory was also employed to calculate the rate constants of-propylbenzene oxidation. The results show that the consumption of-propylbenzene mainly occurs in the alkyl side chain including six C—C and C—H bond fissions of unimolecular reactions and three H-abstraction reactions by O2and other small radicals. Due to the lowest bond dissociation energy, the C—C bond fission adjacent to benzyl radical is the most important consumption channel but the contributions of all H-abstraction reactions are similar. The simulated temperature and density/pressure are positively correlated with the oxidation rate of-propylbenzene, while the effect of equivalence ratio is heavily dependent on the system temperature. Additionally, the calculated apparent activation energies and pre-exponential factors are acceptable compared to the reported experimental results.
-Propylbenzene; Reaction mechanism; High temperature oxidation; ReaxFF; Molecular dynamics simulation
2023-06-10
甯红波, 男, 博士, 副教授, 主要从事燃烧反应动力学研究. E-mail: hbning@swjtu.edu.cn
四川省自然科学基金(批准号: 2023NSFSC1105)、 中央高校基本科研业务费专项资金(批准号: 2682023ZTPY019)和四川省科技厅重点研发计划项目(批准号: 2022YFG0033)资助.
O641
A
10.7503/cjcu20230276
2023-09-04.
Supported by the Natural Science Foundation of Sichuan Province, China(No.2023NSFSC1105), the Fundamental Research Funds for the Central Universities, China(No.2682023ZTPY019) and the Project of Key R&D Program of Sichuan Province, China(No.2022YFG0033).
(Ed.: Y, K, S)
