病原体逃逸宿主固有免疫的新策略:抑制细胞焦亡
2023-11-24杨茂艺崔潇月郑诗棋马诗瑶郑增长邓万燕
杨茂艺,崔潇月,郑诗棋,马诗瑶,郑增长,邓万燕
综 述
病原体逃逸宿主固有免疫的新策略:抑制细胞焦亡
杨茂艺1,崔潇月2,3,郑诗棋2,马诗瑶2,郑增长4,邓万燕2
1. 重庆医科大学感染性疾病分子生物学教育部重点实验室,重庆 400010 2. 重庆医科大学基础医学院病原教研室,重庆 400010 3. 遂宁市中心医院基础实验室,遂宁 629000 4. 同济大学附属东方医院,上海 200120
细胞焦亡是一种由Gasdermin家族蛋白介导的新型程序性细胞死亡。当宿主细胞感应病原体感染或其他危险信号时,Gasdermin家族蛋白被切割活化并诱导细胞焦亡。细胞焦亡过程往往伴随大量炎性细胞因子释放,这些炎性细胞因子在宿主清除病原体过程中发挥着至关重要作用,而病原体在与宿主长期“博弈”过程中也进化出抑制细胞焦亡的策略以实现免疫逃逸。本文介绍了细胞焦亡的发现历程及其在抗感染免疫中的重要功能,并总结了病原体抑制细胞焦亡的多种新策略及其相关研究进展。深入理解细胞焦亡的发生及调控机制,可揭示相关感染性疾病的发病机制并有助于开发有效的抗感染治疗策略。
细胞焦亡;病原体;效应蛋白;免疫逃逸
固有免疫应答是宿主识别与抵抗病原体感染的第一道防线,对激活适应性免疫进而清除病原体至关重要。位于细胞膜表面、内体或细胞质的模式识别受体(pattern recognition receptor,PRRs)通过识别病原体相关分子模式(pathogen-associated molecular patterns,PAMPs)或宿主损伤相关分子模式(damage- associated molecular patterns,DAMPs)启动固有免疫应答抑制病原体的感染[1]。作为固有免疫的重要组成部分,细胞焦亡是一种由Gasdermin (GSDM)家族蛋白活化介导的新型程序性细胞死亡,在清除内源病变或外源感染过程中发挥关键作用[2~4]。1986年,Friedlander等[5]发现炭疽致死毒素(lethal toxin)处理小鼠原代巨噬细胞能够导致细胞死亡和细胞内容物的迅速释放。随后,Black等[6]在1989年报道细胞内存在一种可以剪切白介素1β前体(pro-IL-1β)的天冬氨酸蛋白酶,并将其定义为IL-1β转换酶(IL-1β converting enzyme,ICE)。1992年,Thornberry等[7]首次克隆到ICE基因,并发现ICE蛋白是由p20和p10两个亚基构成的异二聚体。同年,Zychlinsky等[8]发现被福氏志贺氏菌()感染的巨噬细胞存在“自杀”现象。1996年,ICE被重命名为Caspase-1[9]。同年,Chen等[10]进一步发现效应蛋白IpaB (invasion plasmid antigen B)直接结合并活化ICE诱导巨噬细胞发生“炎性细胞凋亡”。Cookson等[11]在2001年首次使用“细胞焦亡”概念来描述这一炎性细胞死亡。Martinon等[12]在2002年首次发现直接激活Caspase-1的蛋白复合物—炎症小体(inflammasome)。2015年,北京生命科学研究所邵峰课题组和美国基因泰克公司Vishva M. Dixit课题组在杂志上背靠背发表了关于细胞焦亡的关键执行蛋白Gasdermin D (GSDMD)的研究成果,成为细胞焦亡研究的里程碑[13,14]。随后研究人员又陆续发现了Gasdermin家族其他成员的内源性活化机制[15],细胞焦亡则因此被重新定义为“由Gasdermin家族蛋白介导的程序性细胞死亡”[13~15]。本文总结了病原体通过抑制炎症小体活化或直接靶向Gasdermin家族蛋白活化等策略抑制细胞焦亡从而逃逸宿主免疫反应的分子机制,讨论了以Gasdermin家族蛋白作为治疗感染性疾病靶点的前景与挑战。
1 细胞焦亡的关键执行蛋白:Gasdermins
Gasdermin家族包含六个成员:GSDMA、GSDMB、GSDMC、GSDMD、GSDME(又称DFNA5)和PJVK(又称DFNB59)[16]。除PJVK外,GSDMA-E均由功能保守的N端结构域(GSDM-NT)和C端结构域(GSDM-CT)组成。GSDM-NT能够结合细胞膜酸性磷脂或线粒体心磷脂,并在膜上多聚化形成孔洞以诱导细胞焦亡[15]。自21世纪初“焦亡”概念提出以来,大量研究结果显示革兰氏阴性菌主要通过激活炎症小体(inflammasome)进而切割活化GSDMD以触发细胞焦亡[13,17~20]。“炎症小体”概念由瑞士洛桑大学生物化学研究所生物医学研究中心Tschopp课题组首次提出,分为经典炎症小体和非经典炎症小体[12]。经典炎症小体主要由Caspase-1、胞质内的模式识别受体(NLRP3、NLRC4、NLRP1、AIM2、Pyrin等)和凋亡相关斑点样蛋白(Apoptosis-associated Speck-like Protein,ASC)组成[21]。经典炎症小体组装后激活Caspase-1进而切割GSDMD,释放GSDMD-NT导致细胞焦亡。与此同时,Caspase-1切割活化IL-1β和白介素18(IL-18)等促炎症因子,招募免疫细胞,清除外源病原体或内源病变[15,22]。非经典炎症小体由病原菌感染条件下释放脂多糖(lipopolysaccharide,LPS)至胞内直接结合并激活Caspase-4/5/11,活化的Caspase-4/5/11切割GSDMD形成具有成孔活性的GSDMD-N端结构域并诱导细胞焦亡[23]。
近年来,多项研究报道了不依赖于炎症小体介导的细胞焦亡通路的活化机制。在抗肿瘤免疫方面,邵峰课题组与美国托马斯杰斐逊大学基梅尔癌症中心生物化学与分子生物学教研室Emad S. Alnemri课题组相继报道细胞凋亡通路的执行蛋白Caspase-3能够切割GSDME,释放GSDME-NT,促使细胞凋亡转变为细胞焦亡[24,25]。此外,美国哈佛大学医学院附属波士顿儿童医院罗鸿博课题组与瑞士病毒学与免疫学研究所Charaf Benarafa 课题组分别报道了GSDMD能够被中性粒细胞弹性蛋白酶(neutrophil elastase,ELANE)和组织蛋白酶G (cathepsin G,CatG)切割活化,引起细胞焦亡[26,27]。美国哈佛大学医学院附属波士顿儿童医院Judy Lieberman课题组报道发现杀伤细胞释放颗粒酶B (granzyme B,GZMB)直接切割肿瘤细胞GSDME触发肿瘤细胞焦亡,提示靶向GSDME是肿瘤免疫疗法的一种潜在的新武器[28]。邵峰课题组发现细胞毒性淋巴细胞分泌的颗粒酶A (granzyme A,GZMA)直接切割肿瘤细胞GSDMB,揭示了GZMA-GSDMB通路介导的焦亡对肿瘤清除的重要性[29]。美国得克萨斯大学安德森癌症中心洪明奇课题组与厦门大学吴乔课题组相继报道Caspase-8剪切GSDMC:一方面,在缺氧的条件下,肿瘤坏死因子(TNF-α)和放线菌酮(cycloheximide,CHX)处理激活了肿瘤细胞Caspase-8,进一步诱导GSMDC的剪切以及细胞焦亡;另一方面,在肿瘤细胞中,代谢产物α-酮戊二酸(α-Ketoglutarate,α-Kg)激活细胞活性氧(reactive oxygen species,ROS)信号导致死亡受体6(death receptor 6,DR6)被氧化,氧化的DR6被内化到胞浆中并招募Caspase-8与GSDMC聚集,随后Caspase-8自激活并切割GSDMC,进一步诱发细胞焦亡[30,31]。在抗感染免疫方面,美国马萨诸塞大学医学院Egil Lien和塔夫茨大学医学院Alexander Poltorak课题组相继发现耶尔森菌()分泌的外膜蛋白J (outer protein J,YopJ)抑制转化生长因子β激活激酶1 (transforming growth factor-β-activated Kinase 1,TAK1),激活RIPK1/Caspase-8复合体,活化的Caspase-8切割GSDMD导致细胞焦亡,但Caspase-8如何被激活还处于未知[32,33]。随后,中国科学院上海免疫与感染研究所刘星课题组通过全基因组CRISPR敲除筛选发现Rag-Ragulator复合体在耶尔森菌感染介导的细胞焦亡中发挥关键作用,揭示了Rag-Ragulator复合体中RagC招募RIPK1和Caspase-8并通过Lamtor1锚定在溶酶体,促使Complex IIb复合体(FADD-RIPK1-Caspase-8)完成组装,RagC GTP酶活性激活Caspase-8触发细胞焦亡[34]。此外,该团队报道了由A族链球菌(Group A,GAS)感染过程中释放的效应蛋白链球菌热源性外毒素B (pyrogenic exotoxin B,SpeB),一种胱氨酸蛋白酶,直接剪切宿主GSDMA诱导细胞焦亡,在限制GAS系统性感染过程中发挥关键作用[35]。这些研究进一步拓展和突出了细胞焦亡在抗感染和抗肿瘤领域的功能。
2 病原体抑制细胞焦亡的新策略
病原体在与宿主长期“博弈”过程中进化出多种抑制细胞焦亡的策略(图1,表1),主要包括:(1)逃逸炎症小体识别;(2)重编程病原体自身代谢抑制炎症小体活化;(3)分泌效应蛋白抑制炎性Caspase的活性;(4)抑制Gasdermin的活化;(5)抑制GSDM-NT的成孔功能。
2.1 逃逸炎症小体识别
现有的报道表明病原体可从抑制模式识别受体激活和表达、抑制炎症小体组装等方面逃逸宿主炎症小体识别:2010年,美国耶鲁大学医学院Ruslan Medzhitov课题组研究发现III型分泌系统(type III secretion system ,T3SS)释放的毒力因子YopK能够与T3SS转位蛋白直接相互作用,阻止宿主细胞对T3SS分泌的效应蛋白的识别,抑制NLRP3炎症小体活化[36];随后,美国石溪大学医学院James B Bliska与美国马萨诸塞大学医学院Egil Lien课题组的研究显示的另一毒力因子YopM能够直接招募并激活宿主激酶PRK1和PRK2,通过磷酸化修饰负调控Pyrin炎症小体活化[37,38]。2017年,香港大学生物医学学院黄建东课题组研究发现金黄色葡萄球菌()分泌的毒力因子腺苷合成酶A (adenosine synthase,AdsA)能够抑制NLRP3的激活,降低巨噬细胞对的杀伤[39]。吉林大学杨勇军课题组报道鼠伤寒沙 门氏菌(serovar Typhimurium,STm)效应蛋白SopB (outer protein B)的肌醇磷酸酶活性在抑制NLRC4的激活中发挥重要作用[40]。德国马克斯·普朗克感染生物学研究所Arturo Zychlinsky课题组的研究表明铜绿假单胞菌(,PA)分泌的绿脓菌素(blue pigment pyocyanin,PCN)通过诱导胞内活性氧和氮氧化物(reactive oxygen species and nitrogen oxides,ROS/RNS)的产生进而抑制NLRP3炎症小体的活化[41]。英国伦敦帝国理工学院Gad Frankel与加拿大麦克马斯特大学Brian K Coombes课题组的研究表明STm 可以下调其鞭毛的表达,以逃避NLRC4的识别,而甲型副伤寒沙门氏菌(,SPtA)产生超长的LPS O-抗原链,能够干扰多种炎症小体的激活[42,43]。2023年,美国哈佛大学医学院Benjamin E Gewurz课题组的最新研究表明EB病毒(Epstein-Barr virus,EBV)能够通过类泛素化修饰其编码的G蛋白偶联受体BILF1以抑制线粒体抗病毒信号蛋白(mitochondrial antiviral-signaling protein,MAVS)介导的NLRP3炎症小体活化[44]。美国新墨西哥大学Vojo Deretic课题组在2008年研究发现结核分枝杆菌(MTB)通过NLRC4激活Caspase-1,这一过程被锌金属蛋白酶(Zn2+metalloprotease,ZMP1)所抑制,但ZMP1如何作用的确切细节尚不清楚[45]。美国北卡罗来纳大学莱恩伯格综合癌症中心Blossom Damania课题组的研究表明卡波西肉瘤相关疱疹病毒(Kaposi’s sarcoma-associated herpesvirus,KSHV) ORF63蛋白与人NLRP1同源,该蛋白能够直接与NLRP1相互作用,通过抑制NLRP1寡聚化以及NLRP1与Pro-caspase-1结合来阻止炎症小体的组装[46]。根据美国马里兰大学医学院Joao HF Pedra课题组的报道,在立克次体()感染过程中,蜱虫唾液中的效应蛋白唾液抑素L2与磷脂结合蛋白膜联蛋白A2结合阻断NLRC4炎症小体寡聚化,抑制了下游Caspase-1的激活以及细胞因子的释放[47]。日本爱知医科大学医学院Takashi Yokochi课题组发现仙台病毒(Sendai Virus) V蛋白通过与NLRP3相互作用,抑制NLRP3-ASC的结合和随后ASC寡聚从而破坏NLRP3炎症小体复合物的组装[48]。墨西哥国立理工大学Vianney Ortiz-Navarrete课题组研究发现STmT3SS可通过诱导转录共激活因子Yes相关蛋白(yes-associated protein,YAP)磷酸化及其与造血细胞激酶(hematopoietic cell kinase,HCK)的相互作用来阻止YAP易位到细胞核,p73/YAP不再结合到NLRC4启动子区,导致NLRC4表达下调[49]。苏州大学吴淑燕课题组发现了STm质粒毒力因子C (Plasmid Virulence C,SpvC)通过下调NLRP3和NLRC4的表达以抑制炎症小体活化并加剧全身性感染[50]。干扰宿主炎症小体对病原体的识别已经成为细菌发生免疫逃逸的新策略,对于增强细菌的毒力并建立有效的感染至关重要。靶向该免疫逃逸策略的治疗方案的开发将有助于在固有免疫应答早期限制感染的程度,阻止病原体扩散。
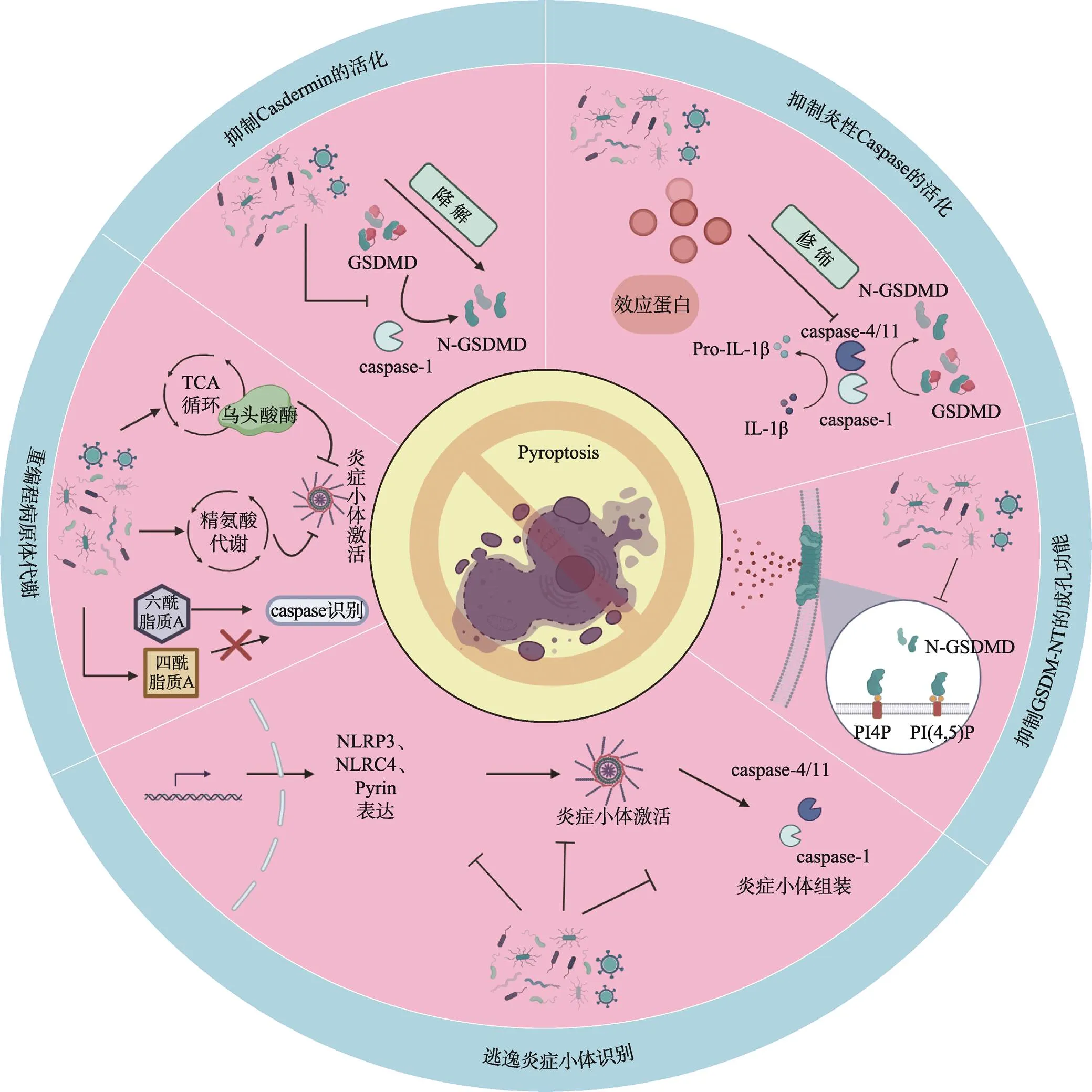
图1 病原体抑制细胞焦亡的新策略
图绘制网址:https://www.biorender.com/。
2.2 重编程病原体代谢
华东理工大学阳大海课题组发现在感染巨噬细胞的过程中,杀鱼爱德华氏菌()激活自身精氨酸代谢途径,招募宿主细胞的多胺转运系统,将细胞质内的精氨酸转化成精胺,导致宿主细胞质内精胺积累,阻断钾离子外流,抑制NLRP3炎症小体的激活[51]。美国宾夕法尼亚大学Igor E. Brodsky课题组通过转座子突变库筛选发现STmTCA循环中介导NLRP3炎症小体免疫逃逸的基因——乌头酸酶,该基因的缺失导致NLRP3炎症小体的快速激活,提示通过操纵氧化代谢逃逸NLRP3炎症小体[52]。美国加州大学David M. Underhill课题组发现通过化学修饰其细胞壁组分肽聚糖(PGN),使其能够抵御溶酶体的消化,抑制NLRP3炎症小体激活以及破坏IL-1β的分泌[53]。LPS的结构中脂质A能够被Caspase-11所识别,而病原菌通过不同的策略改变LPS脂质A结构逃逸这一过程。 Vinogradov等[54]与Hagar等[55]的研究显示新凶手弗朗西斯菌()通过乙酰化修饰脂质A,逃避了Caspase-11的识别,而脂质A能够发生脱酰化形成Caspase无法识别的四酰脂质A。未来的研究将致力于探寻能够调控代谢以发生免疫逃避的病原体,并确定是否涉及其他细菌代谢物。针对该免疫逃逸策略的进一步研究可以为开发基于病原体代谢系统抑制剂的抗菌策略开辟道路。
2.3 抑制炎性Caspase的活化
多项研究显示病原体分泌的效应蛋白能够直接与Caspase相互作用并抑制其活性从而阻断炎症小体介导的细胞焦亡。美国耶鲁大学医学院Richard A Flavell课题组研究发现T3SS分泌的细胞外酶U (exoenzyme U,ExoU)通过其A2磷脂酶活性抑制Caspase-1的激活[56]。Jakka等[57]发现布鲁氏菌()编码的一种含TIR结构域的效应蛋白(TIR domain-containing protein encoded by Brucella,TcpB )能够与Caspase-1/4/11相互作用并促进Caspase-1/4/11的泛素化和降解,抑制Caspase-1/4/11介导的炎症信号转导。英国帝国理工学院Gad Frankel课题组发现肠致病性大肠杆菌(Enteropathogenic Escherichia coli,EPEC)和肠出血性大肠杆菌(Enterohemorrhagic E. coli,EHEC)T3SS效应蛋白non-LEE encoded effector F(NleF)直接结合并通过干扰Caspase-4/11不同亚基的多聚化从而抑制其蛋白酶活性[58,59]。日本东京大学Chihiro Sasakawa课题组发现福氏志贺菌()外膜蛋白C3(outer Shigella proteins C3,OspC3)也具有与NleF类似的作用机制[60]。邵峰课题组进一步研究揭示了OspC3通过ADP-核糖基化修饰Caspase-4/11并使其失活来实现对非经典炎症小体活化的抑制[61]。此外,英国剑桥大学Felix Randow和邵峰课题组相继研究发现侵袭质粒抗原H (invasion plasmid antigen H 9.8,IpaH9.8)通过泛素化并降解鸟苷酸结合蛋白1-4 (guanylate binding proteins 1-4,GBP1-4),使得病原体被限制在囊泡内,无法将LPS释放到胞质中[62,63]。圣保罗大学Dario S. Zamboni课题组发现由伯氏考克斯体() Dot/ICM IV型分泌系统分泌的效应蛋白—半胱天冬酶活性抑制蛋白A (inhibition of Caspase activation,IcaA)同样能够抑制Caspase-11的激活[64]。美国康涅狄格大学Sivapriya Kailasan Vanaja课题组研究显示EHEC噬菌体编码的毒力因子志贺毒素(toxin,STX)能够抑制LPS诱导的非经典炎症小体的激活,且该过程依赖于STX2的催化活性[65]。但上述两项研究的具体作用机制尚不清晰。分泌毒力因子抑制炎症性Caspase 的活化是病原体免疫逃逸的重要策略,发掘具有该逃逸策略的病原体并进一步明确其逃逸机制将为宿主-病原体相互作用的研究以及相关感染性疾病的治疗提供新的见解。
2.4 抑制Gasdermin的活化
Gasdermin家族蛋白是细胞焦亡的关键执行蛋白,病原体可以直接抑制该家族蛋白的活性和表达而抑制细胞焦亡[66,67]。北京大学夏朋延课题组的研究表明新型冠状病毒(SARS-CoV-2)核衣壳能够直接结合GSDMD-NT和GSDMD-CT之间的连接区,保护GSDMD免受Caspase-1的切割从而抑制细胞焦亡[66]。中国医学科学院北京协和医学院王建伟课题组研究发现肠病毒71型(enterovirus,EV71)分泌的蛋白酶3C能够识别并切割GSDMD的Q193位点,剪切所形成的GSDMD-N端结构域不具备成孔功能,从而抑制细胞焦亡[67]。美国基因泰克公司Vishva M. Dixit课题组研究表明T3SS效应蛋白IpaH7.8能够识别并泛素化人源的GSDMD使其被蛋白酶体降解[68]。此外,美国德克萨斯大学Neal M. Alto课题组使用泛素激活的相互作用捕获技术(ubiquitin- activated interaction traps,UBAITs)证明IpaH7.8同样识别宿主GSDMB,能够通过降解GSDMB有效阻止自然杀伤细胞清除上皮细胞中的[69,70]。上述研究丰富了病原体对抗宿主防御系统的机制,针对该免疫逃逸机制的研究对靶向病原体分泌蛋白的抑制剂的开发具有重要价值。
2.5 抑制GSDM-NT的成孔功能
泛素化是一种能够调节多种细胞功能的翻译后修饰,由活化酶 E1,结合酶E2和连接酶E3催化完成[71]。泛素化不仅能够靶向底物蛋白以26S 蛋白酶体依赖的方式使其降解,还在调节先天性和适应性免疫反应以及免疫耐受方面具有关键作用。一些病原体已进化出复杂的机制以逃避或抵消泛素依赖的宿主反应,其中由病原体编码的E3泛素连接酶和去泛素化酶在病原体-宿主细胞相互作用中发挥至关重要的作用[72,73]。中国科学院微生物研究所刘翠华团队的研究表明MTB能够通过劫持泛素来改变宿主膜的磷脂组成以抑制细胞焦亡:MTB分泌的蛋白质酪氨酸磷酸酶B (protein tyrosine phosphatase B,PtpB)通过泛素结合依赖的方式使宿主细胞膜磷脂酰肌醇-4-磷酸(PI4P)和磷脂酰肌醇-(4,5)-二磷酸[PI(4,5)P2]去磷酸化,导致 GSDM-NT无法在细胞膜上成孔从而抑制细胞焦亡,促进其在巨噬细胞内的存活[74]。泛素系统介导的宿主-病原体相互作用为抗感染治疗提供了潜在的选择。
3 结语与展望
细胞焦亡是机体抵抗病原体感染的一种重要天然免疫反应,而病原体在宿主免疫系统的压力下进化出了适应性策略来抑制细胞焦亡以实现持续感染。本综述重点总结了病原体(包括病原菌和病毒)抑制细胞焦亡的研究进展,并讨论了病原体及其表达的毒力因子通过逃逸炎症小体识别或阻止炎症小体的组装与激活、降解Gasdermin家族蛋白以及破坏其细胞膜成孔功能等方式抑制宿主细胞焦亡发生的机制。
病原体逃逸细胞焦亡的相关研究有助于进一步了解病原体-宿主相互作用机制,为开发新型抗感染治疗策略奠定了重要理论基础。这些研究为感染性疾病提供几种潜在的治疗策略:(1)通过靶向病原体分泌系统的抑制剂,破坏其逃逸细胞焦亡效应因子的释放来限制病原体的感染[76]。近年来,针对病原体T3SS系统的抑制剂发展迅速。T3SS复合物的关键作用蛋白PcrV已在动物模型中得到有效验证可以作为的免疫预防靶标[77]。目前正在研发的抗PcrV制剂(KB001)已进入IIa期临床试验[78,79];此外,PcrV/Psl双特异性人单克隆抗体MEDI3902目前处于IIb期临床试验阶段[80];(2)通过在感染过程中特异性激活细胞焦亡实现对病原体的清除[81]。有研究表明在脓毒症早期,细胞焦亡具有保护性作用[82,83],适当的细胞焦亡可通过破坏被感染的细胞促进病原体的释放从而募集免疫细胞来激活宿主免疫反应,促进病原体的清除和机体对感染的抵抗。有研究表明,在脓毒症早期进行谷氨酰胺的预防性给药可以促进肝细胞焦亡从而增强对病原体的清除效果[84];(3)细胞焦亡的代谢调控为感染性疾病的治疗提供新的前景[85],基于在感染过程中由病原体自身或感染的宿主代谢重编程特点,通过改变相应代谢途径或代谢物以促进细胞焦亡来控制病原体的感染。尽管如此,这些策略在临床中的应用仍面临巨大挑战,如正常细胞被异常激活细胞焦亡从而导致炎性因子风暴。
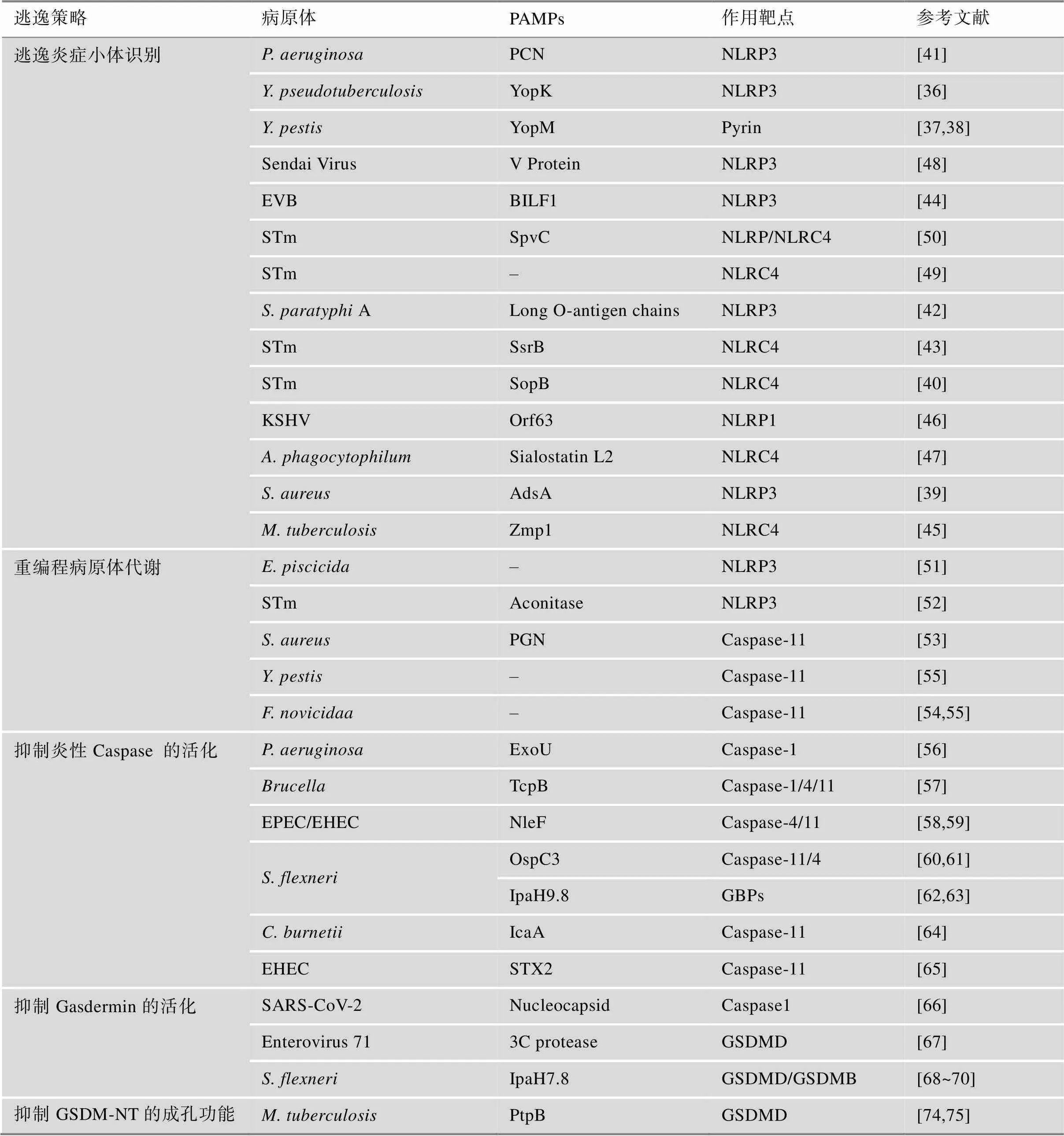
表1 病原体抑制细胞焦亡的新策略
虽然人们在发现病原体应对宿主防御机制的对策或逃避策略方面已取得显著进展,但对于这一领域的认知尚不够深入,可能存在其他未知的逃逸宿主细胞焦亡的病原体或机制。未来的研究应继续致力于发掘具有逃逸宿主细胞焦亡的病原体并阐明细胞内病原体的免疫逃逸机制,为靶向宿主细胞焦亡的药物开发提供新思路,并为设计新型疫苗和治疗方案以应对感染性疾病奠定重要基础。
[1] Kanneganti TD. Intracellular innate immune receptors: life inside the cell, 2020, 297(1): 5–12.
[2] Man SM, Karki R, Kanneganti TD. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases, 2017, 277(1): 61–75.
[3] Man SM, Kanneganti TD. Regulation of inflammasome activation, 2015, 265(1): 6–21.
[4] Jorgensen I, Miao EA. Pyroptotic cell death defends against intracellular pathogens, 2015, 265(1): 130–142.
[5] Friedlander AM. Macrophages are sensitive to anthrax lethal toxin through an acid-dependent process, 1986, 261(16): 7123–7126.
[6] Black RA, Kronheim SR, Merriam JE, March CJ, Hopp TP. A pre-aspartate-specific protease from human leukocytes that cleaves pro-interleukin-1 beta, 1989, 264(10): 5323–5326.
[7] Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, Miller DK, Molineaux SM, Weidner JR, Aunins J. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes, 1992, 356(6372): 768–774.
[8] Zychlinsky A, Prevost MC, Sansonetti PJ. Shigella flexneri induces apoptosis in infected macrophages, 1992, 358(6382): 167–169.
[9] Alnemri ES, Livingston DJ, Nicholson DW, Salvesen G, Thornberry NA, Wong WW, Yuan J. Human ICE/CED-3 protease nomenclature, 1996, 87(2): 171.
[10] Chen Y, Smith MR, Thirumalai K, Zychlinsky A. A bacterial invasin induces macrophage apoptosis by binding directly to ICE, 1996, 15(15): 3853–3860.
[11] Cookson BT, Brennan MA. Pro-inflammatory programmed cell death, 2001, 9(3): 113–114.
[12] Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflamematory caspases and processing of proIL-beta, 2002, 10(2): 417–426.
[13] Shi JJ, Zhao Y, Wang K, Shi XY, Wang Y, Huang HW, Zhuang YH, Cai T, Wang FC, Shao F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death, 2015, 526(7575): 660–665.
[14] Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, Liu PS, Lill JR, Li H, Wu JS, Kummerfeld S, Zhang J, Lee WP, Snipas SJ, Salvesen GS, Morris LX, Fitzgerald L, Zhang YF, Bertram EM, Goodnow CC, Dixit VM. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling, 2015, 526(7575): 666–671.
[15] Shi JJ, Gao WQ, Shao F. Pyroptosis: Gasdermin-mediated programmed necrotic cell death, 2017, 42(4): 245–254.
[16] Zheng ZZ, Deng WY, Lou XW, Bai Y, Wang JH, Zeng HS, Gong ST, Liu X. Gasdermins: pore-forming activities and beyond, 2020, 52(5): 467–474.
[17] Ding JJ, Wang K, Liu W, She Y, Sun Q, Shi JJ, Sun HZ, Wang DC, Shao F. Pore-forming activity and structural autoinhibition of the gasdermin family, 2016, 535(7610): 111–116.
[18] Liu X, Zhang ZB, Ruan JB, Pan YD, Magupalli VG, Wu H, Lieberman J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores, 2016, 535(7610): 153–158.
[19] Hu JJ, Liu X, Xia SY, Zhang ZB, Zhang Y, Zhao JX, Ruan JB, Luo XM, Lou XW, Bai Y, Wang JH, Hollingsworth LR, Magupalli VG, Zhao L, Luo HR, Kim J, Lieberman J, Wu H. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation, 2020, 21(7): 736–745.
[20] Xia SY, Zhang Z, Magupalli VG, Pablo JL, Dong Y, Vora SM, Wang LF, Fu TM, Jacobson MP, Greka A, Lieberman J, Ruan JB, Wu H. Gasdermin D pore structure reveals preferential release of mature interleukin-1, 2021, 593(7860): 607–611.
[21] Kesavardhana S, Malireddi RKS, Kanneganti TD. Caspases in cell death, inflammation, and pyroptosis, 2020, 38: 567–595.
[22] Liu X, Lieberman J. A mechanistic understanding of pyroptosis: the fiery death triggered by invasive infection, 2017, 135: 81–117.
[23] Shi JJ, Zhao Y, Wang YP, Gao WQ, Ding JJ, Li P, Hu LY, Shao F. Inflammatory caspases are innate immune receptors for intracellular LPS, 2014, 514(7521): 187–192.
[24] Wang YP, Gao WQ, Shi XY, Ding JJ, Liu W, He HB, Wang K, Shao F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin, 2017, 547(7661): 99–103.
[25] Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death, 2017, 8: 14128.
[26] Kambara H, Liu F, Zhang XY, Liu P, Bajrami B, Teng Y, Zhao L, Zhou SY, Yu HB, Zhou WD, Silberstein LE, Cheng T, Han MZ, Xu YF, Luo HR. Gasdermin D exerts anti-inflammatory effects by promoting neutrophil death, 2018, 22(11): 2924–2936.
[27] Burgener SS, Leborgne NGF, Snipas SJ, Salvesen GS, Bird PI, Benarafa C. Cathepsin G inhibition by Serpinb1 and Serpinb6 prevents programmed necrosis in neutrophils and monocytes and reduces GSDMD-driven inflammation, 2019, 27(12): 3646–3656.e5.
[28] Zhang ZB, Zhang Y, Xia SY, Kong Q, Li SY, Liu X, Junqueira C, Meza-Sosa KF, Mok TMY, Ansara J, Sengupta S, Yao Y, Wu H, Lieberman J. Gasdermin E suppresses tumour growth by activating anti-tumour immunity, 2020, 579(7799): 415–420.
[29] Zhou ZW, He HB, Wang K, Shi XY, Wang YP, Su Y, Wang Y, Li D, Liu W, Zhang YL, Shen LJ, Han WD, Shen L, Ding JJ, Shao F. Granzyme a from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells, 2020, 368(6494): eaaz7548.
[30] Hou JW, Zhao RC, Xia WY, Chang CW, You Y, Hsu JM, Nie L, Chen Y, Wang YC, Liu CX, Wang WJ, Wu Y, Ke BZ, Hsu JL, Huang KB, Ye Z, Yang Y, Xia XH, Li YT, Li CW, Shao B, Tainer JA, Hung MC. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis, 2020, 22(10): 1264–1275.
[31] Zhang JY, Zhou B, Sun RY, Ai YL, Cheng K, Li FN, Wang BR, Liu FJ, Jiang ZH, Wang WJ, Zhou DW, Chen HZ, Wu Q. The metabolite alpha-KG induces GSDMC-dependent pyroptosis through death receptor 6-activated caspase-8, 2021, 31(9): 980–997.
[32] Sarhan J, Liu BC, Muendlein HI, Li P, Nilson R, Tang AY, Rongvaux A, Bunnell SC, Shao F, Green DR, Poltorak A. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection, 2018, 115(46): E10888–E10897.
[33] Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, Brooks A, Xia SY, Wu H, Kelliher MA, Berger SB, Gough PJ, Bertin J, Proulx MM, Goguen JD, Kayagaki N, Fitzgerald KA, Lien E. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death, 2018, 362(6418): 1064–1069.
[34] Zheng ZZ, Deng WY, Bai Y, Miao R, Mei SL, Zhang ZB, Pan YD, Wang Y, Min R, Deng F, Wu ZY, Li W, Chen PC, Ma TC, Lou XW, Lieberman J, Liu X. The lysosomal Rag-Ragulator complex licenses RIPK1 and Caspase-8- mediated pyroptosis by Yersinia, 2021, 372(6549): eabg0269.
[35] Deng WY, Bai Y, Deng F, Pan YD, Mei SL, Zheng ZZ, Min R, Wu ZY, Li W, Miao R, Zhang ZB, Kupper TS, Lieberman J, Liu X. Streptococcal pyrogenic exotoxin B cleaves GSDMA and triggers pyroptosis, 2022, 602(7897): 496–502.
[36] Brodsky IE, Palm NW, Sadanand S, Ryndak MB, Sutterwala FS, Flavell RA, Bliska JB, Medzhitov R. A Yersinia effector protein promotes virulence by preventing inflammasome recognition of the type III secretion system, 2010, 7(5): 376–387.
[37] Chung LK, Park YH, Zheng YT, Brodsky IE, Hearing P, Kastner DL, Chae JJ, Bliska JB. The Yersinia virulence factor YopM hijacks host kinases to inhibit Type III effector-triggered activation of the Pyrin inflammasome, 2016, 20(3): 296–306.
[38] Ratner D, Orning MP, Proulx MK, Wang D, Gavrilin MA, Wewers MD, Alnemri ES, Johnson PF, Lee B, Mecsas J, Kayagaki N, Goguen JD, Lien E. The Yersinia pestis Effector YopM inhibits Pyrin inflammasome activation, 2016, 12(12): e1006035.
[39] Deng J, Zhang BZ, Chu H, Wang XL, Wang YX, Gong HR, Li RH, Yang D, Li C, Dou Y, Gao P, Cai JP, Jin ML, Du Q, Chan JFW, Kao RYT, Yuen KY, Huang JD. Adenosine synthase A contributes to recurrent Staphylococcus aureus infection by dampening protective immunity, 2021, 70: 103505.
[40] Hu GQ, Song PX, Chen W, Qi S, Yu SX, Du CT, Deng XM, Ouyang HS, Yang YJ. Cirtical role for Salmonella effector SopB in regulating inflammasome activation, 2017, 90: 280–286.
[41] Virreira Winter S, Zychlinsky A. The bacterial pigment pyocyanin inhibits the NLRP3 inflammasome through intracellular reactive oxygen and nitrogen species, 2018, 293(13): 4893–4900.
[42] Mylona E, Sanchez-Garrido J, Hoang Thu TN, Dongol S, Karkey A, Baker S, Shenoy AR, Frankel G. Very long O-antigen chains of Salmonella Paratyphi A inhibit inflammasome activation and pyroptotic cell death, 2021, 23(5): e13306.
[43] Ilyas B, Mulder DT, Little DJ, Elhenawy W, Banda MM, Pérez-Morales D, Tsai CN, Chau NYE, Bustamante VH, Coombes BK. Regulatory evolution drives evasion of host inflammasomes by Salmonella typhimurium, 2018, 25(4): 825–832.e5.
[44] Yiu SPT, Zerbe C, Vanderwall D, Huttlin EL, Weekes MP, Gewurz BE. An Epstein-Barr virus protein interaction map reveals NLRP3 inflammasome evasion via MAVS UFMylation, 2023, 83(13): 2367–2386.e15.
[45] Master SS, Rampini SK, Davis AS, Keller C, Ehlers S, Springer B, Timmins GS, Sander P, Deretic V. Mycobacterium tuberculosis prevents inflammasome activation, 2008, 3(4): 224–232.
[46] Gregory SM, Davis BK, West JA, Taxman DJ, Matsuzawa S, Reed JC, Ting JP, Damania B. Discovery of a viral NLR homolog that inhibits the inflammasome, 2011, 331(6015): 330–334.
[47] Wang XW, Shaw DK, Sakhon OS, Snyder GA, Sundberg EJ, Santambrogio L, Sutterwala FS, Dumler JS, Shirey KA, Perkins DJ, Richard K, Chagas AC, Calvo E, Kopecky J, Kotsyfakis M, Pedra JHF. The Tick protein Sialostatin L2 binds to Annexin A2 and inhibits NLRC4-mediated inflammasome activation, 2016, 84(6): 1796–1805.
[48] Komatsu T, Tanaka Y, Kitagawa Y, Koide N, Naiki Y, Morita N, Gotoh B, Yokochi T. Sendai virus V protein inhibits the secretion of interleukin-1beta by preventing NLRP3 inflammasome assembly, 2018, 92(19): e00842–18.
[49] Perez-Lopez A, Rosales-Reyes R, Alpuche-Aranda CM, Ortiz-Navarrete V. Salmonella downregulates Nod-like receptor family CARD domain containing protein 4 expression to promote its survival in B cells by preventing inflammasome activation and cell death, 2013, 190(3): 1201–1209.
[50] Zuo LL, Zhou LT, Wu CY, Wang YL, Li YY, Huang R, Wu SY. Salmonella spvC gene inhibits pyroptosis and intestinal inflammation to aggravate systemic infection in mice, 2020, 11: 562491.
[51] Jiang JT, Wang WW, Sun F, Zhang YX, Liu Q, Yang DH. Bacterial infection reinforces host metabolic flux from arginine to spermine for NLRP3 inflammasome evasion, 2021, 34(10): 108832.
[52] Wynosky-Dolfi MA, Snyder AG, Philip NH, Doonan PJ, Poffenberger MC, Avizonis D, Zwack EE, Riblett AM, Hu B, Strowig T, Flavell RA, Jones RG, Freedman BD, Brodsky IE. Oxidative metabolism enables Salmonella evasion of the NLRP3 inflammasome, 2014, 211(4):653–668.
[53] Shimada T, Park BG, Wolf AJ, Brikos C, Goodridge HS, Becker CA, Reyes CN, Miao EA, Aderem A, Götz F, Liu GY, Underhill DM. Staphylococcus aureus evades lysozyme-based peptidoglycan digestion that links phagocytosis, inflammasome activation, and IL-1beta secretion, 2010, 7(1): 38–49.
[54] Vinogradov E, Perry MB, Conlan JW. Structural analysis of Francisella tularensis lipopolysaccharide, 2002, 269(24): 6112–6118.
[55] Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock, 2013, 341(6151): 1250–1253.
[56] Sutterwala FS, Mijares LA, Li L, Ogura Y, Kazmierczak BI, Flavell RA. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome, 2007, 204(13): 3235–3245.
[57] Jakka P, Namani S, Murugan S, Rai N, Radhakrishnan G. The Brucella effector protein TcpB induces degradation of inflammatory caspases and thereby subverts non-canonical inflammasome activation in macrophages, 2017, 292(50): 20613–20627.
[58] Pallett MA, Crepin VF, Serafini N, Habibzay M, Kotik O, Sanchez-Garrido J, Di Santo JP, Shenoy AR, Berger CN, Frankel G. Bacterial virulence factor inhibits caspase-4/11 activation in intestinal epithelial cells, 2017, 10(3): 602–612.
[59] Song T, Li KW, Zhou W, Zhou J, Jin Y, Dai HM, Xu TT, Hu MD, Ren HG, Yue JJ, Liang L. A Type III effector NleF from EHEC inhibits epithelial inflammatory cell death by targeting Caspase-4, 2017, 2017: 4101745.
[60] Kobayashi T, Ogawa M, Sanada T, Mimuro H, Kim M, Ashida H, Akakura R, Yoshida M, Kawalec M, Reichhart JM, Mizushima T, Sasakawa C. The Shigella OspC3 effector inhibits caspase-4, antagonizes inflammatory cell death, and promotes epithelial infection, 2013, 13(5): 570–583.
[61] Li ZL, Liu W, Fu JQ, Cheng S, Xu Y, Wang ZQ, Liu XF, Shi XY, Liu YX, Qi XB, Liu XY, Ding JJ, Shao F. Shigella evades pyroptosis by arginine ADP-riboxanation of caspase-11, 2021, 599(7884): 290–295.
[62] Wandel MP, Kim BH, Park ES, Boyle KB, Nayak K, Lagrange B, Herod A, Henry T, Zilbauer M, Rohde J, MacMicking JD, Randow F. Guanylate-binding proteins convert cytosolic bacteria into caspase-4 signaling platforms, 2020, 21(8): 880–891.
[63] Li P, Jiang W, Yu Q, Liu W, Zhou P, Li J, Xu JJ, Xu B, Wang FC, Shao F. Ubiquitination and degradation of GBPs by a Shigella effector to suppress host defence, 2017, 551(7680): 378–383.
[64] Cunha LD, Ribeiro JM, Fernandes TD, Massis LM, Khoo CA, Moffatt JH, Newton HJ, Roy CR, Zamboni DS. Inhibition of inflammasome activation by Coxiella burnetii type IV secretion system effector IcaA, 2015, 6: 10205.
[65] Havira MS, Ta A, Kumari P, Wang CL, Russo AJ, Ruan JB, Rathinam VA, Vanaja SK. Shiga toxin suppresses noncanonical inflammasome responses to cytosolic LPS, 2020, 5(53): eabc0217.
[66] Ma, J, Zhu FR, Zhao M, Shao F, Yu D, Ma JW, Zhang XS, Li WT, Qian Y, Zhang Y, Jiang D, Wang S, Xia P. SARS-CoV-2 nucleocapsid suppresses host pyroptosis by blocking Gasdermin D cleavage, 2021, 40(18): e108249.
[67] Lei XB, Zhang ZZ, Xiao X, Qi JL, He B, Wang JW. Enterovirus 71 inhibits pyroptosis through cleavage of Gasdermin D, 2017, 91(18): e01069–17.
[68] Luchetti G, Roncaioli JL, Chavez RA, Schubert AF, Kofoed EM, Reja R, Cheung TK, Liang YX, Webster JD, Lehoux I, Skippington E, Reeder J, Haley B, Tan MW, Rose CM, Newton K, Kayagaki N, Vance RE, Dixit VM. Shigella ubiquitin ligase IpaH7.8 targets gasdermin D for degradation to prevent pyroptosis and enable infection, 2021, 29(10): 1521–1530.e10.
[69] Hansen JM, de Jong MF, Wu Q, Zhang LS, Heisler DB, Alto LT, Alto NM. Pathogenic ubiquitination of GSDMB inhibits NK cell bactericidal functions, 2021, 184(12): 3178–3191.e18.
[70] Yin H, Zheng J, He QQ, Zhang X, Li XZC, Ma YJ, Liang X, Gao JQ, Kocsis BL, Li Z, Liu X, Alto NM, Li L, Zhang H. Insights into the GSDMB-mediated cellular lysis and its targeting by IpaH7.8, 2023, 14(1): 61.
[71] Scheffner M, Nuber U, Huibregtse JM. Protein ubiquitination involving an E1-E2-E3 enzyme ubiquitin thioester cascade, 1995, 373(6509): 81–83.
[72] Hu HB, Sun SC. Ubiquitin signaling in immune responses, 2016, 26(4): 457–483.
[73] Mukherjee R, Dikic I. Regulation of host-pathogen interactions via the ubiquitin system, 2022, 76: 211–233.
[74] Chai QY, Yu SS, Zhong YZ, Lu Z, Qiu CG, Yu Y, Zhang XW, Zhang Y, Lei ZH, Qiang LH, Li BX, Pang Y, Qiu XB, Wang J, Liu CH. A bacterial phospholipid phosphatase inhibits host pyroptosis by hijacking ubiquitin, 2022, 378(6616): eabq0132.
[75] Krause DS, Dikic I. Mycobacterium tuberculosis hijacks ubiquitin to inhibit pyroptosis, 2022, 82(24): 4588–4590.
[76] Zigangirova NA, Nesterenko LN, Sheremet AB, Soloveva AV, Luyksaar SI, Zayakin ES, Balunets DV, Gintsburg AL. Fluorothiazinon, a small-molecular inhibitor of T3SS, suppresses salmonella oral infection in mice, 2021, 74(4): 244–254.
[77] Warrener P, Varkey R, Bonnell JC, DiGiandomenico A, Camara M, Cook K, Peng L, Zha JY, Chowdury P, Sellman B, Stover CK. A novel anti-PcrV antibody providing enhanced protection against Pseudomonas aeruginosa in multiple animal infection models, 2014, 58(8): 4384–4391.
[78] Jain R, Beckett VV, Konstan MW, Accurso FJ, Burns JL, Mayer-Hamblett N, Milla C, VanDevanter DR, Chmiel JF. KB001-A, a novel anti-inflammatory, found to be safe and well-tolerated in cystic fibrosis patients infected with Pseudomonas aeruginosa, 2018, 17(4): 484–491.
[79] Milla CE, Chmiel JF, Accurso FJ, VanDevanter DR, Konstan MW, Yarranton G, Geller DE. Anti-PcrV antibody in cystic fibrosis: a novel approach targeting Pseudomonas aeruginosa airway infection., 2014, 49(7): 650–658.
[80] Ali SO, Yu XQ, Robbie GJ, Wu Y, Shoemaker K, Yu L, DiGiandomenico A, Keller AE, Anude C, Hernandez-Illas M, Bellamy T, Falloon J, Dubovsky F, Jafri HS. Phase 1 study of MEDI3902, an investigational anti–Pseudomonas aeruginosa PcrV and Psl bispecific human monoclonal antibody, in healthy adults., 2019, 25(5): 629.e1–629.e6.
[81] Xue YS, Enosi Tuipulotu D, Tan WH, Kay C, Man SM. Emerging activators and regulators of inflammasomes and pyroptosis., 2019, 40(11): 1035–1052.
[82] Garnacho-Montero J, Palacios-García I, Díaz-Martín A, Gutiérrez-Pizarraya A, López-Sánchez JM, Gómez EA, Cordero MD. Sequential changes of NLRP3 inflammasome activation in sepsis and its relationship with death., 2020, 54(3): 294–300.
[83] Huet O, Pickering RJ, Tikellis C, Latouche C, Long F, Kingwell B, Dickinson B, Chang CJ, Masters S, Mackay F, Cooper ME, de Haan JB. Protective effect of inflamemasome activation by hydrogen peroxide in a mouse model of septic shock., 2017, 45(2): e184–e194.
[84] Pai MH, Wu JM, Yang PJ, Lee PC, Huang CC, Yeh SL, Lin MT. Antecedent dietary glutamine supplementation benefits modulation of liver pyroptosis in mice with polymicrobial sepsis., 2020, 12(4): 1086.
[85] Place DE, Kanneganti TD. Metabolic regulation of pyroptotic cell death expands the therapeutic landscape for treating inflammatory disease, 2021, 6(1): 37.
Inhibiting pyroptosis: novel immune evasion strategies for pathogens
Maoyi Yang1, Xiaoyue Cui2,3, Shiqi Zheng2, Shiyao Ma2, Zengzhang Zheng4, Wanyan Deng2
Pyroptosis is a type of programmed cell death mediated by the Gasdermin family. It is triggered in response to pathogen infection or other danger signals. The activation of Gasdermins leads to pyroptosis and the release of large amounts of inflammatory cytokines. Pyroptosis plays a crucial role in combating pathogen infections, as it helps to eliminate infected cells and activate the immune system. However, pathogens have already developed sophisticated strategies to evade or inhibit pyroptosis, allowing them to persist and facilitate infection. This review provides an overview of the discovery of pyroptosis and its importance in anti-infectious immunity. We also discuss several new strategies for inhibiting pyroptosis by pathogens. A thorough learning of the occurrence and regulation of pyroptosis may reveal the pathogenesis of related infectious diseases and contribute to developing effective anti-infective therapeutic strategies.
pyroptosis; pathogen; effector protein; immune evasion
2023-07-18;
2023-10-18;
2023-11-02
重庆市自然科学基金创新发展联合基金(编号:CSTB2022NSCQ-LZX0031)资助[Sponsored by the Natural Science Foundation of Chongqing, China (No. CSTB2022NSCQ-LZX0031)]
杨茂艺,硕士研究生,专业方向:临床检验诊断学。E-mail: yang9910150028@163.com
邓万燕,博士,研究员,研究方向:宿主-病原体相互作用。E-mail: dengwanyan@cqmu.edu.cn
郑增长,博士,副研究员,研究方向:天然免疫激活与调控。E-mail: zhengzengzhang@126.com
10.16288/j.yczz.23-191
(责任编委: 谢建平)
