硫化钼催化剂用于催化多环芳烃芘的加氢反应*
2023-11-22王悦燚贾永梁马晓迅
王悦燚 王 婧 贾永梁 马晓迅 孙 鸣
(西北大学化工学院,碳氧资源清洁利用国家国际科技合作基地,陕北能源先进化工利用技术 教育部工程研究中心,陕西省洁净煤转化工程技术研究中心,陕北能源化工产业 发展协同创新中心,710069 西安)
0 引 言
由于原油产量不能完全满足生产需求,同时石油资源日益减少[1-2],因此,众多学者作出了不懈努力,开发出了多种可利用能源,以弥补原油的不足。我国能源国情为缺油、少气、富煤,清洁高效地利用煤炭转化副产的焦油,是对石油资源的重要补充。对煤焦油中重质组分的不完全利用会导致碳资源的浪费[3],因此许多研究人员对煤焦油,尤其是高温煤焦油进行了进一步研究。国内外对煤焦油的利用主要分为组分提纯和加氢精制[4]。德国吕特格公司从高温煤焦油中分离出更具经济价值的蒽、萘、咔唑和沥青等组分,且综合利用率位居世界首位[5]。俄罗斯研究人员将高温煤焦油中多种多环芳烃通过催化加氢反应制备成高密度喷气燃料的原料,以获得燃料馏分[6]。国内的煤焦油利用主要是按照温度蒸馏切割后对各馏分进行精制以及对煤焦油进行加氢[7]。对高温煤焦油进行蒸馏切割后可以得到不同温度段的馏分,其中温度高于360 ℃时的沥青馏分中富含多环芳烃[7],具备多元的利用价值。在已有的文献报道中,多环芳烃的应用途径有很多。蒽由于其对称性被广泛用作合成一些以蒽为前体的双光子吸收材料,在光信息储存、有机发光二极管等领域有重大作用[8];菲可以合成菲醌,作为农作物杀虫剂、工业染料、光导材料等[9-10];芘可以作为原材料制备抗原,用以研究水产品中芘的残留量,还可以作为太阳能电池中空穴传输材料的合成原料之一[11-12]。多环芳烃加氢后还可以生成环烷烃,将环烷烃混合到喷气燃料中,可以提高体积热值和燃烧性能[6,13]。
目前,针对萘、蒽、菲的催化加氢研究已非常成熟[14-17],而对四环芳烃芘的研究较鲜见。芘催化加氢是多路径的反应过程[18],加氢产物多达7种,而低饱和加氢产物逐步加氢并最终达到饱和,以此得到的加氢产物——环烷基油品具有良好的热沉性能及较高的能量密度和吸热性等优点。在催化加氢研究中选择较多的催化剂为贵金属催化剂[18-19],虽然催化剂活性高,但因成本高、不易回收、抗硫性较差等问题而使应用受到限制。相反,过渡金属催化剂则可在添加助剂后改善其催化性能,且具有成本较低、来源广泛等优点,因此,在中低温煤焦油及萘、蒽、菲等多环芳烃的催化加氢反应中被广泛应用和研究。过渡金属硫化物如MoS2,具有典型的二维层状结构,层间通过较弱的范德华力连接,层内由较强的Mo—S共价键连接,沿层内Mo—S键的棱面化学性质比较活泼[20],可作为催化反应的活性位点,尤其是在催化加氢反应研究中运用广泛。芘作为大分子直径达到1.2 nm[21],而催化剂载体大多使用传统的分子筛和金属氧化物,孔径较小,如传统Al2O3载体孔道小且与活性组分有较强相互作用[22];当TiO2和ZrO2作为载体时,反应大部分在催化剂表面进行[23]。载体MCM-41等虽然具备了可调节孔径大小的优点,但由于一维孔道容易堵塞,不利于反应[24]。而泡沫镍具有三维大孔结构,孔径可达到几百微米[25],更有利于活性物种的分散和大分子反应[23]。
为进一步探讨多环芳烃化合物的催化加氢反应,本研究首次使用大孔三维泡沫镍作为催化剂载体,采用水热合成法制备了以非贵金属钼为活性组分、硫为助剂的钼基催化剂,应用于高温煤焦油高温馏分中的多环芳烃芘的催化加氢反应中,通过改变催化加氢反应时的温度、压力、时间以及催化剂质量和活性组分比例等条件,考察了催化剂的催化加氢性能,研究了助剂硫提高钼基催化剂催化反应活性的作用机制,探讨了非贵金属钼基催化剂催化加氢反应机理。
1 实验部分
1.1 原料与试剂
本实验采用的原料与试剂有:质量分数为99.0%的四水合钼酸铵((NH4)6Mo7O24·4H2O),由广州金华大化学试剂有限公司生产;质量分数为99.0%的硫脲(CH4N2S)、质量分数为99.0%的尿素(CO(NH2)2)、质量分数为96.0%的氟化铵(NH4F)、质量分数为99.7%的环己烷(C6H12),均由天津大茂化学试剂厂生产;质量分数为97%的芘(C16H10),由上海麦克林生化科技公司生产;480 g/cm3泡沫镍,由合肥科晶材料技术有限公司生产。
1.2 催化剂制备
先将2 cm×2 cm的泡沫镍(NF)放入3 mol/mL的盐酸溶液中超声预处理,再依次用无水乙醇和蒸馏水进行冲洗,随后烘干备用。
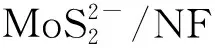
1.3 催化剂的表征方法
采用德国Bruker公司生产的D8 Advance型X射线衍射仪对催化剂晶相结构进行X射线衍射(XRD)分析,Cu靶,扫描速率为2°/min,扫描范围为20°~70°。
采用德国ZEISS公司生产的Sigma 3000型扫描电子显微镜(SEM)对催化剂形貌、表面组分等进行测试,测试时加速电压为3 kV,能谱Mapping拍摄时加速电压为15 kV,探测器为SE2二次电子探测器。
采用美国FEI公司生产的F200X型透射电子显微镜(TEM)对催化剂的形貌和微观结构进行表征,工作电压为200 kV。
采用美国Thermo Scientific公司生产的X射线光电子能谱测量催化剂表面物质的结合能,用AlK α辐射作为激发源,以C1s=284.8 eV为标准进行荷电校准。
1.4 催化剂的芘加氢性能评价方法
本研究在250 mL不锈钢高温高压反应釜中进行,通过程序升温评价Mo基催化剂的催化加氢反应性能。反应物为1 g模型化合物芘,溶剂为34 g环己烷。在反应温度为200 ℃~340 ℃,反应压力为5 MPa~6 MPa条件下,催化加氢反应2 h后进行取样分析。另外,芘转化率采用气相色谱-质谱联用仪测定,按照面积归一化法计算。
经传输管道将需要测试的样品输送至日本岛津公司生产的QP2010 plus型气相色谱-质谱联用仪(GC-MS)中进行在线分析,传输管道及进样阀温度设置为300 ℃。色谱柱为Rxi-5 ms毛细管柱(30 m×0.25 mm×0.25 μm),采用分流模式,分流比为1∶80。GC升温程序为:首先将温度升至60 ℃,保持1 min;随后以10 ℃/min的升温速率由60 ℃升温至90 ℃保持1 min后,再以10 ℃/min的升温速率由90 ℃升温至170 ℃保持1 min;最后以10 ℃/min由170 ℃升至300 ℃,保持10 min。热解气的化学组成通过参考NIST图谱库和已知的煤焦油组分进行确定。
2 结果与讨论
2.1 XRD表征
采用XRD对Mo基催化剂的晶型结构进行表征分析,结果如图1所示。由图1可以看出,催化剂在2θ为44.5°(111)和51.8°(200)两处的衍射峰来自于催化剂的载体泡沫镍。负载金属后,催化剂在2θ为21.7°,31.1°,37.7°,49.7°,50.1°和55.2°处出现了衍射峰,分别对应于Ni3S2的(101),(110),(003),(113),(211)和(122)晶面,说明添加的助剂硫与泡沫镍载体发生了反应,载体中部分镍被消耗,形成新的Ni3S2晶型。另外,图1中没有出现活性物种钼的特征衍射峰,说明钼物种可能均匀地分散于泡沫镍载体上。
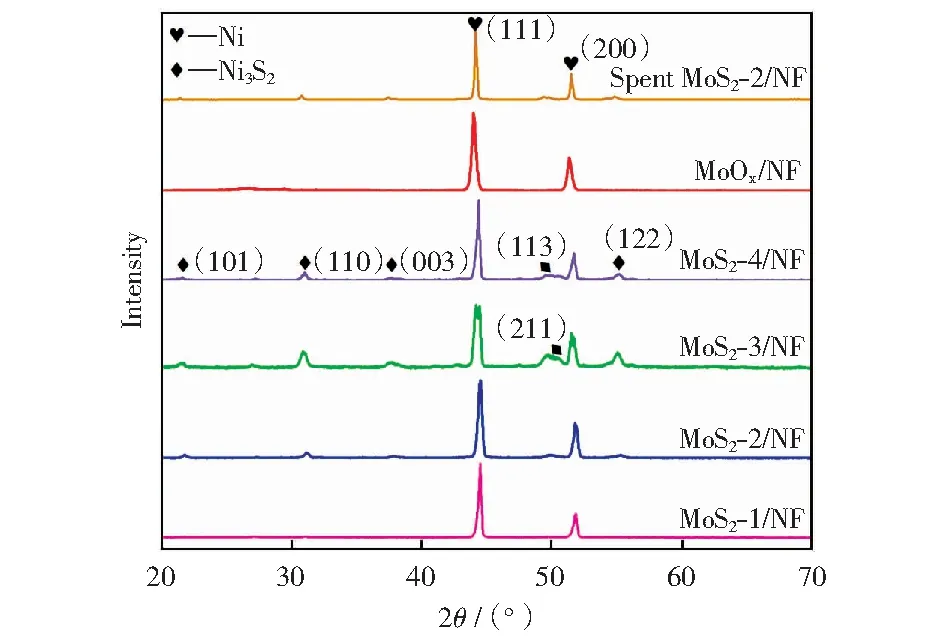
图1 Mo基催化剂的XRD谱Fig.1 XRD patterns of Mo-based catalysts
2.2 SEM表征
图2所示为Mo基催化剂的SEM照片。由图2a可以看出,MoOx/NF催化剂具有花瓣状纳米片结构,壁厚约为20 nm,呈现出超薄特性,这是纳米颗粒定向生长的结果[20],导致催化剂在催化加氢反应中活性较高。由图2b和图2d可以看出,硫的掺杂使得钼物种在泡沫镍表面出现了团聚现象,而图2c和图2f中的催化剂表面却呈现出不同的纳米片状结构,相对于未掺硫的催化剂,其纳米片结构分布不是很均匀,活性物种的团聚解释了XRD中Ni衍射峰强度变弱的现象。另外,Spent MoS2-2/NF催化剂的纳米片厚度显著增加,可能的原因是反应后催化剂表面的活性物质发生了团聚,片状结构明显变厚,导致XRD表征中Ni的衍射峰发生偏移。
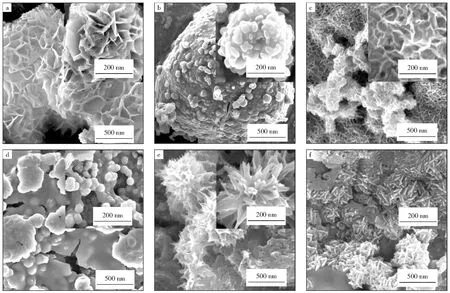
图2 Mo基催化剂的SEM照片Fig.2 SEM photos of Mo-based catalysts a—MoOx/NF;b—MoS2-1/NF;c—MoS2-2/NF;d—MoS2-3/NF;e—MoS2-4/NF;f—Spent MoS2-2/NF
图3所示为Mo基催化剂的Mapping照片。由图3可以看出,元素Mo和S都均匀分布在催化剂表面上。表1所示为Mo基催化剂的Mapping结果。由表1可以看出,随着硫脲添加量增加,S元素的质量分数呈现先升高后降低的趋势,推测原因是硫脲添加量过高影响活性物种和载体之间的相互作用力,在水热过程中发生脱落。由此可知,掺硫钼物种成功地负载在泡沫镍载体上,这与XRD结果一致,因此,掺硫钼物种良好的分散性是MoS2-2/NF催化剂转化率提高的主要原因之一。
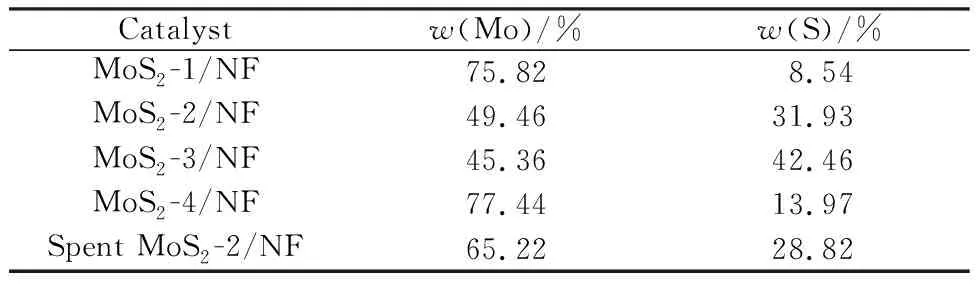
表1 Mo基催化剂的Mapping结果Table 1 Mapping results of Mo-based catalysts
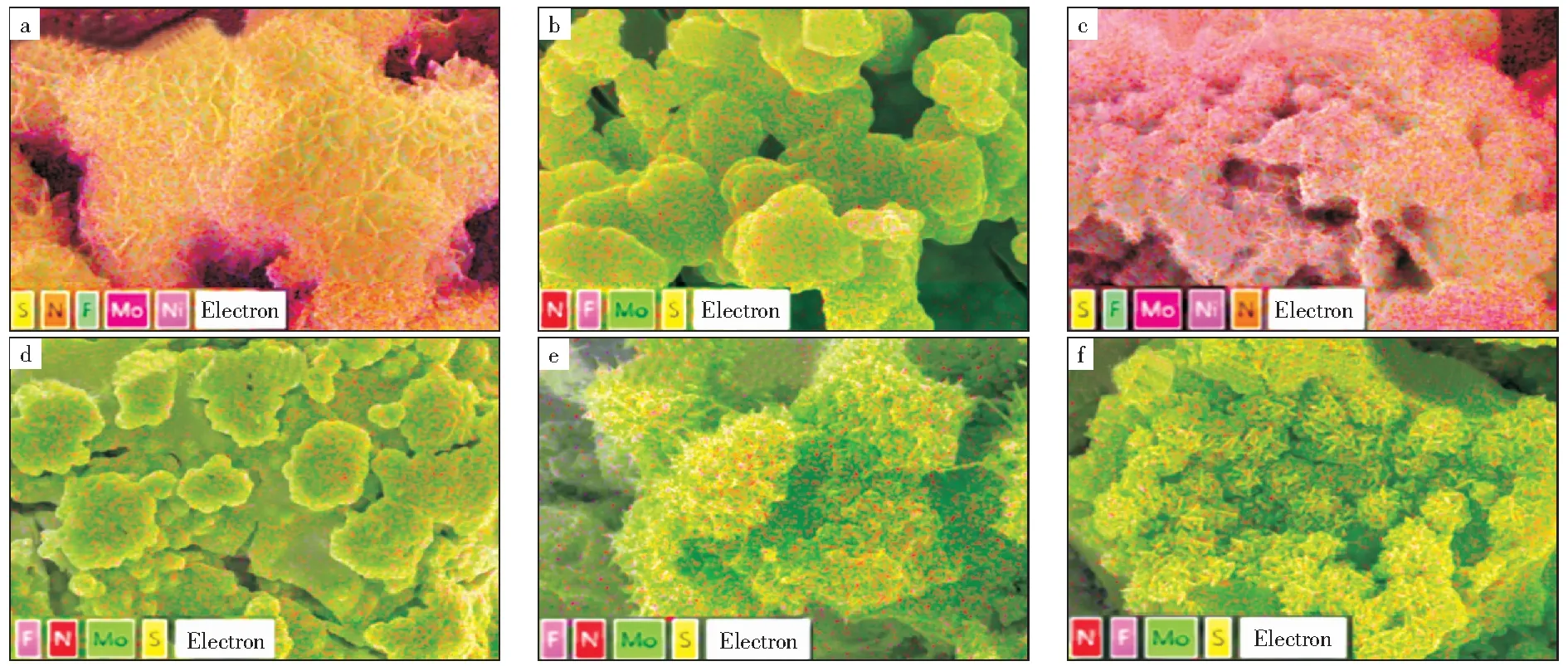
图3 Mo基催化剂的Mapping照片Fig.3 Mapping photos of Mo-based catalysts a—MoOx/NF;b—MoS2-1/NF;c—MoS2-2/NF;d—MoS2-3/NF;e—MoS2-4/NF;f—Spent MoS2-2/NF
对MoS2-2/NF催化剂进行TEM表征分析以确定催化剂的结构和晶型,结果如图4所示。由图4可以清晰观察到,催化剂表面颗粒大小约为8 nm,呈现纳米片层状结构,相互交叠,生长在载体泡沫镍上[26],进一步证实了催化剂为层状结构,堆积密度较小,纳米片层数为5~10,长度约为5 nm。纳米尺寸越小,暴露的棱边越多,活性位点越多,提高了催化剂的催化性能。由图4b可知,0.62 nm的晶格间距对应MoS2的(002)晶面,证明催化剂的活性物种为MoS2。

图4 MoS2-2/NF催化剂的TEM照片Fig.4 TEM photos of MoS2-2/NF
2.3 XPS表征
为探究Mo基催化剂表面Mo物种的价态组成,对MoS2-2/NF催化剂样品进行了XPS分析表征(见图5)。图5a所示为反应前催化剂Mo 3d的XPS谱。由图5a可以看出,结合能在232.1 eV处的峰归属于Mo 3d3/2,对应Mo4+的硫化态MoS2,因此,硫的掺杂有利于活性物种MoS2的形成[27]。结合能在235.7 eV和232.6 eV处的峰分别归属于Mo 3d3/2和Mo 3d5/2,对应Mo6+的氧化态MoO3[28-29],高价态的MoO3活性物种有利于催化剂的催化性能提高。
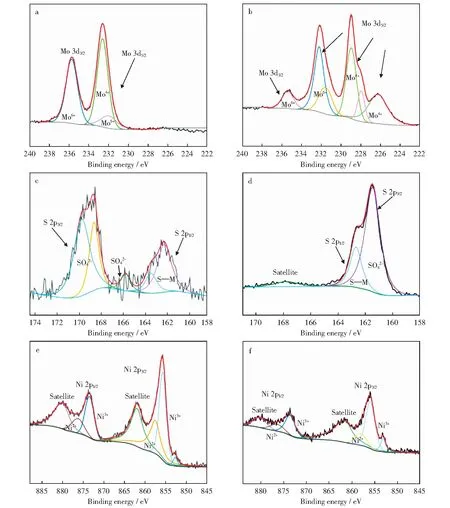
图5 MoS2-2/NF催化剂的XPS谱 Fig.5 XPS spectra of MoS2-2/NF a—Mo 3d XPS spectra of MoS2-2/NF;b—Mo 3d XPS spectra of Spent MoS2-2/NF;c—S 2p XPS spectra of MoS2-2/NF;d—S 2p XPS spectra of Spent MoS2-2/NF;e—Ni 2p XPS spectra of MoS2-2/NF;f—Ni 2p XPS spectra of Spent MoS2-2/NF
图5b所示为反应后催化剂Mo 3d的XPS谱。由图5b可以看出,反应后的催化剂在235.4 eV,232.6 eV和231.7 eV结合能处的峰分别归属于Mo 3d3/2,Mo 3d5/2,Mo 3d5/2,对应Mo6+的氧化态MoO3[28-29],在229 eV处和228 eV处的峰对应Mo4+[29],说明反应后催化剂的活性物种没有发生改变。此外,在226.3 eV结合能处的峰归属于氧化态MoO2中的Mo4+[28]。因此,催化剂中的部分MoO3活性组分可能在催化加氢反应过程中被还原,导致反应活性在循环反应中下降。

图5e所示为反应前催化剂Ni 2p的XPS谱,可以拟合为两个自旋轨道双子星和两个卫星峰。由图5e可以看出,结合能在872.9 eV和855.2 eV处的峰分别归属于Ni 2p1/2和Ni 2p3/2,对应Ni3S2中的Ni3+[32,34],这与XRD表征结果一致,并且Ni3S2具备加氢活性[29],有利于进一步提高催化剂的加氢活性。另外,结合能在875.4 eV和856.8 eV处的峰分别归属于Ni 2p1/2和Ni 2p3/2,对应NiOx中的Ni2+[32,37],而结合能在879.9 eV和862 eV处的峰分别归属于Ni 2p1/2和Ni 2p3/2,是两个自旋轨道的卫星峰[37]。其中,活性物种Ni3S2和NiOx均来自于水热过程中泡沫镍载体的消耗,而泡沫镍载体属于惰性载体,可以被活化的Ni元素很少。因此,对于芘催化加氢反应影响不大,后续以金属Mo的活性物种为主要活性物种进行讨论。
图5f所示为反应后催化剂Ni 2p的XPS谱。由图5f可以看出,反应前后的Ni 2p XPS谱基本一致。在873.2 eV和855.7 eV处的峰对应Ni3S2中的Ni3+,与反应前结合能相差0.5 eV以内,与XRD中镍物种反应后没有改变这一结论相符。结合能在875.9 eV和857.3 eV处的峰对应NiOx中的Ni2+。峰面积和反应前催化剂Ni 2p XPS谱的峰面积相比有所减少,可能是由于反应过程中催化剂表面上镍物种的脱落。
2.4 芘催化加氢的反应结果
不同反应条件对MoOx/NF催化剂的影响如图6所示。
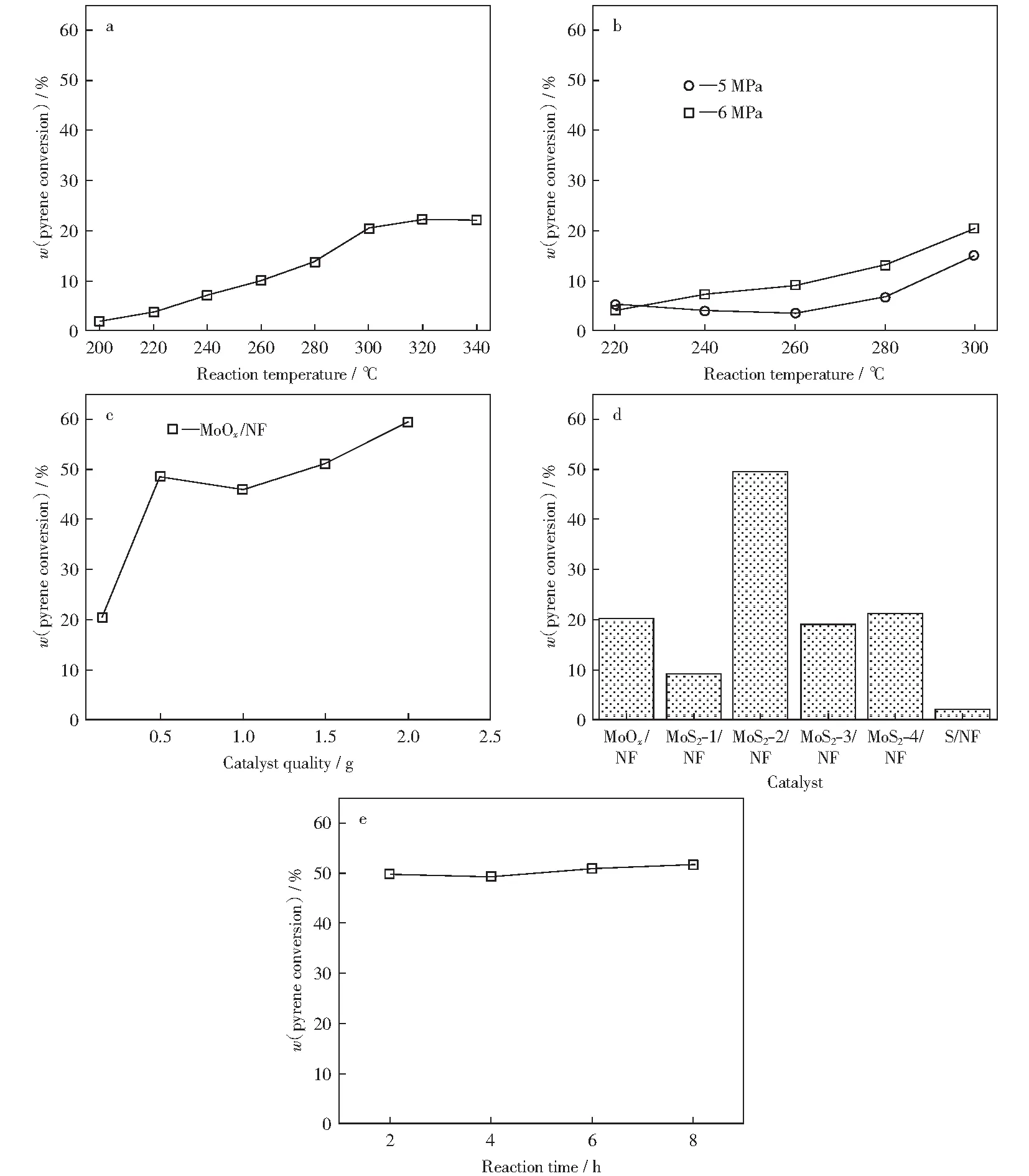
图6 不同反应条件对MoOx/NF催化剂催化性能的影响Fig.6 Effect of different reaction conditions on catalytic performance of MoOx/NF catalyst a—Reaction temperature;b—Reaction pressure;c—Catalyst quality;d—Sulfur doping amounts;e—Reaction time
2.4.1 反应温度对催化剂性能的影响
根据文献[1],设定反应压力为5 MPa,反应时间为2 h,催化剂质量为0.15 g,在反应温度为200 ℃~340 ℃的范围内优化出最佳反应温度。由图6a可知,MoOx/NF催化剂的催化活性随着反应温度升高而增大,即芘转化率呈上升趋势。由图6a还可知,在反应温度为300 ℃时,芘转化率为20.38%,随后芘转化率上升趋势逐渐平缓,因此,选取300 ℃为较优反应温度。
2.4.2 反应压力对催化剂性能的影响
在反应温度为220 ℃~300 ℃,反应时间为2 h,催化剂质量为0.15 g,反应压力分别为5 MPa和6 MPa条件下,通过对比催化剂的催化活性,优化出适合反应的反应压力。由图6b可以看出,相较于反应压力为5 MPa时的反应活性,催化剂在反应压力为6 MPa时反应活性更高,即芘转化率更高。因此,在保证催化加氢反应安全进行的前提下,优化出的催化加氢反应压力为6 MPa。
2.4.3 催化剂质量对催化剂性能的影响
通过对比实验,将一组泡沫镍载体在水热时不添加前驱体进行反应,焙烧后称量质量的变化,结果发现,水热反应前载体质量为0.212 g,水热反应并焙烧后质量变为0.209 g,质量差可以忽略。因为催化剂载体质量较大,实际通过ICP测得负载量为4%(质量分数)左右,因此分别选取催化剂质量为0.15 g,0.50 g,1.00 g和2.00 g,在筛选出的反应温度300 ℃,反应压力6 MPa的条件下进行实验,结果如图6c所示。由图6c可以看出,随着催化剂质量增加,芘转化率呈现增加趋势。值得注意的是,当催化剂用量为0.5 g时,芘转化率增加最快,由原来的20.38%增加到了48.49%。但随着催化剂质量继续增加,虽然芘转化率有所上升,但增幅较小,因此,选取最佳催化剂质量为0.5 g。
2.4.4 硫元素的掺杂量对催化剂性能的影响
煤焦油中含有硫的杂环化合物使催化剂在实际加氢过程中容易中毒失活,掺硫可以提供更多空位,改善催化剂性能,并且二维纳米片状MoS2边缘位点具有类金属的连续电子结构,可提升催化剂活性[38]。
由图6d可知,当硫脲添加量为2 mmol时,对应MoS2-2/NF催化剂,表现出最好的催化活性,即芘转化率最高,约为49.54%。表2所示为不同硫掺杂量催化剂的芘转化率及产物选择性。由表2可知,MoS2-2/NF催化剂深度加氢产物(1,2,3,6,7,8-六氢芘(C):17.18%,1,2,3,3a,4,5-六氢芘(D):3.19%)的选择性达到了20.37%。相比较而言,不含硫的MoOx/NF催化剂的芘转化率仅为20.38%,说明硫的掺杂提供了更多活性位点,有效提高了催化剂的催化加氢反应活性。另外,只含硫不含活性金属钼的S/NF催化剂几乎没有活性,因此,高活性MoS2物种有效提高了催化剂的催化加氢反应活性,这与FU et al[21]的研究结果一致。相较于MoS2-1/NF,MoS2-3/NF,MoS2-4/NF催化剂,MoS2-2/NF催化剂性能更好的主要原因可能是,在SEM表征中该催化剂表现出超薄的片状结构和均匀的活性物种分布,证明助剂硫脲的添加量过多或过少都不利于活性物种分布,影响活性物种和载体之间的相互作用力,分布不均匀、颗粒过大均会导致催化剂稳定性下降。

表2 不同硫掺杂量催化剂的芘转化率及产物选择性Table 2 Pyrene conversions and products selectivities of catalysts with different sulfur doping amounts
2.4.5 反应时间对催化剂性能的影响
选取芘转化率最高的MoS2-2/NF催化剂,考察反应时间对催化加氢反应活性的影响。本实验选取前期考察出的反应条件,分别测试了加氢反应在反应时间为2 h,4 h,6 h,8 h时的催化剂活性,结果如图6e所示。由图6e可知,随着反应时间增加,催化剂活性并没有显著提升,反应在2 h时芘转化率已经达到较高,在8 h时活性基本不变,因此,考虑到时间和成本等问题,反应时间选取2 h。
2.5 催化剂稳定性考察
选取最佳反应条件(反应温度为300 ℃,氢气压力为6 MPa,催化剂质量为0.5 g,反应时间为2 h),进行催化剂循环使用实验,结果如图7所示。由图7可知,催化剂使用两次后芘转化率由49.54%降至24.78%;催化剂使用三次后,芘转化率降至6.23%,表明制备的催化剂稳定性差。在XPS表征中,反应后的催化剂有部分MoO3中的Mo6+还原至MoO2中的Mo4+,说明在催化加氢反应中钼物种部分被还原,并且在SEM表征中发现泡沫镍载体表面的活性物种发生团聚,造成催化剂性能降低。因此,在马弗炉中350 ℃焙烧2 h后进行再生,改善催化剂的稳定性。在马弗炉中对使用过三次的MoS2-2/NF催化剂进行焙烧,考察其对芘催化加氢反应的催化性能,结果如表3所示。由表3可以看出,再生后的催化剂的芘转化率上升至38.27%,证明催化剂具有可再生性,后续将对进一步提高催化剂的稳定性开展研究。
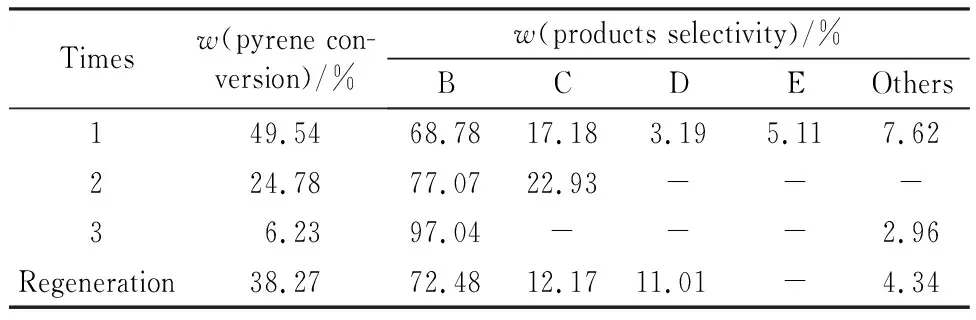
表3 催化剂稳定性Table 3 Stability of MoS2-2/NF catalyst
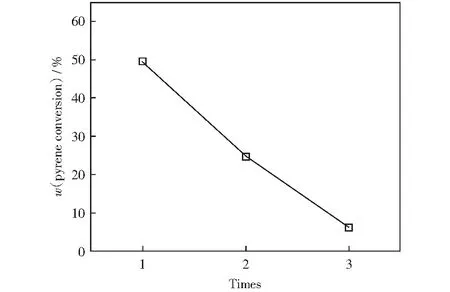
图7 MoS2-2/NF催化剂稳定性曲线Fig.7 Stability curve of MoS2-2/NF catalyst
3 结 论
1) 负载在泡沫镍载体上的活性物种MoS2具有超薄的纳米片状结构,且具有良好的分散性,在载体上均匀分布。
2) 助剂硫的添加使得催化剂产生硫空位,从而生成更多的活性位点。超薄的纳米片状结构有效提高了催化剂加氢反应活性。
3) 当反应温度为300 ℃,氢气压力为6 MPa,反应时间为2 h,催化剂质量为0.5 g,硫脲添加量为2 mmol时,MoS2-2/NF催化剂表现出最佳的催化活性,对比MoOx/NF催化剂,多环芳烃化合物芘转化率由原来的20.38%增加至49.54%,其中深度加氢产物1,2,3,3a,4,5-六氢芘和1,2,3,6,7,8-六氢芘选择性为20.37%。
4) 对反应后的MoS2-2/NF催化剂进行再生,转化率可以达到38.27%,证明催化剂具有可再生性。
