超高效液相色谱-Orbitrap 高分辨质谱联用快速筛查测定鱼虾贝中79 种药物残留
2023-11-20姜萌艺汤志旭罗忻赵元晖王雷付晓婷许加超高昕
姜萌艺,汤志旭,罗忻,赵元晖,王雷,付晓婷,许加超,高昕*
(1.中国海洋大学 食品科学与工程学院,山东 青岛 266003;2.青岛海关技术中心,山东 青岛 266109)
近年来,随着大食物观提出,鱼虾类水产品愈发受到消费者的青睐。2021 年我国养殖水产品产量达到5 394.41 万t,约占水产品总产量的81%[1]。兽药在水产养殖中发挥着防治疾病、促进生长、提高品质等重要作用[2]。由于兽药本身对生物和自然环境的危害以及其沿食物链在生物体内积聚的特性[3-4],许多国家和地区都对水产品中兽药使用及最大残留限量进行了规定[5-6]。我国农业农村部第250 号公告中明确规定了21 类禁用兽药,GB 31650—2019《食品安全国家标准食品中兽药最大残留限量》[7]中规定了食品中104 种兽药的最大残留限量,其中有22 种药物规定了在鱼类中的限量值。欧盟、美国、日本也分别在相关法规中规定了121、87、651 种兽药在水产品中的最大残留限量[8-9]。
2023 年1 月1 日新实行的《中华人民共和国农产品质量安全法》第十三条规定:国家建立农产品质量安全风险监测制度,国务院农业农村主管部门应当制定国家农产品质量安全风险监测计划。目前我国水产品中兽药残留检测方法相关的国家标准和行业标准[10-11],主要针对河豚鱼、鳗鱼和烤鳗等水生产品及其相应的加工品种中的单个药残品类,缺少包含多种类水产品及其水产加工品的检测方法,且这些方法大部分是基于液相色谱串联质谱技术,其检测的兽药化合物种类少且单一[12-14],亟需建立能够针对多种类水产品中多种兽药残留同时检测的简单、高效、灵敏度高的方法[15-17]。基于此,本研究利用超高效液相色谱-Orbitrap 高分辨质谱技术[high performance liquid chromatography-high resolution orbitrap mass spectrometry,UPLC-HRMS(Orbitrap)],在对比正负离子模式下各兽药化合物响应强度的基础上构建高分辨质谱信息库,并采用多种有机溶剂提取和固相萃取净化的前处理方法,建立能够对鱼虾贝类水产品中的11 类79 种兽药残留的同时快速筛查检测方法,以期为水产品中多种兽药残留检测提供有力的理论依据和技术支撑。
1 材料与方法
1.1 材料与试剂
乙腈、甲醇(色谱级):德国Merck 公司;甲酸、乙酸(质谱级):美国Sigma-Aldrich 公司;甲酸铵、乙酸铵(色谱级):美国Mallinckrodt Baker 公司;乙二胺四乙酸二钠盐(ethylenediaminetetraacetic acid disodium salt,EDTA-2Na,分析纯):国药集团化学试剂有限公司;Oasis HLB 固相萃取柱(3cc/60 mg)、Acquity UPLC BEH C18(100 mm×2.1 mm×1.7 μm):美国Waters 公司;MEGA BE-C18固相萃取柱(1 GM/6 mL):美国Agilent 公司;0.22 μm 针式滤膜过滤头:天津市津腾实验设备有限公司。
79 种兽药标准品(纯度>95%)、长尾鳕(Macrouroidei)、南美白对虾(Litopenaeus Vannamei)、栉孔扇贝(Azumapecten farreri):市售。
1.2 仪器与设备
Dionex UltiMate 3000 UHPLC+超高压液相色谱仪、Q Exactive Focus 质谱仪:美国Thermo Scientific 公司;Milli-Q 超纯水仪:美国Millipore 公司;CR21G 高速冷冻离心机:日本HITACHI 公司;MS3 Basic 旋涡振荡器:德国IKA 公司;MMV-1000W 垂直振荡器:日本EYELA 公司;PL303 电子天平:瑞士Mettler Toledo 公司;TurboVap LV 氮吹仪:美国Caliper 公司。
1.3 标准溶液
分别准确称取11 类79 种兽药化合物25.0 mg于25 mL 容量瓶内制成浓度为1 mg/mL 标准储备液,再按11 种类别用甲醇配制成浓度为10 μg/mL 混合标准品中间液,于-20 ℃避光条件下保存。β-内酰胺类化合物的稳定性较差,需要单独配制避光保存;配制标准储备液时β-内酰胺类使用纯净水定容,磺胺及甲氧苄啶类、三苯甲烷染料类使用乙腈定容,其余化合物使用甲醇定容。试验时,将上述混合标准品中间液使用甲醇配制成所需浓度的混合标准工作液。
1.4 仪器条件
1.4.1 液相色谱条件
色谱柱:Acquity UPLC BEH C18色谱柱(100 mm×2.1 mm×1.7 μm);进样量:2 μL;柱温:35 ℃;流速:0.3 mL/min;流动相A 为2 mmol/L 甲酸铵(含0.01%甲酸水溶液),B 为甲醇。洗脱程序:0~0.5 min,5% B;0.5~12.0 min,5%~95%B;12.0~16.0 min,95%B;16.1~20.0 min,5%~95%B。
1.4.2 质谱条件
离子源为加热电喷雾离子源(heat electron spray ionization,HESI),毛细管温度为320 ℃,喷雾电压分别为3.5 kV(正离子模式)和2.9 kV(负离子模式);透镜电压为50 V;鞘气流速为2.758×105Pa;辅助气温度为300 ℃。对正负离子同时进行采集,数据采集模式采用全扫描/数据依赖的二级扫描模式(full mass-data dependent scans,Full MS-dd MS2)方式,根据目标化合物的质量范围(m/z),将扫描范围设置为140.0~1 000.0;一级质谱分辨率[以50%峰宽计算,即测定一个峰半峰高处的全峰宽(full width half maximum,FWHN)]为70 000 FWHN,自动增益控制目标离子数(automatic gain control target,AGC target) 为1×106,C-trap 最大注入时间为100 ms;二级质谱分辨率为17 500 FWHM,dd-MS2AGC target 为2×105,C-trap 最大注入时间为50 ms;归一化碰撞能(normalized collision energy,NCE)为20%、40%、60%;动态排除时间为60 s。
1.5 样品制备及前处理
取待检测的长尾鳕、南美白对虾、栉孔扇贝样品,其中长尾鳕样品去骨,南美白对虾样品去头,栉孔扇贝样品开壳,分别取可食用部分切块绞碎混合均匀后,于-20 ℃条件下保存备用。
称取5.0 g(精确至0.01 g)试样于50 mL 聚丙烯离心管中,静置,向其中加入10 mmol/L EDTA-2Na 溶液0.5 mL 后,再加入混合提取溶液[乙腈∶甲醇∶水(3∶1∶1,体积比,含1%乙酸)]25 mL,均质提取5 min,在4 °C下12 000 r/min 离心15 min,取5 mL 上清液,于40 ℃条件下氮气吹干,甲醇定容至1.0 mL。将提取液负载至Oasis HLB 固相萃取柱(使用前用1 mL 水、1 mL 甲醇预处理)上,保持1 滴/s 的流速,依次用1 mL 水洗涤,1 mL 甲醇洗脱,收集全部流出液,40 °C 氮气吹干,用甲醇定容至1.0 mL,涡旋混匀,过0.22 μm 微孔滤膜后上机测试。
优化试验以长尾鳕为基质,采用空白添加的试验方法,以回收率作为评判指标,向提取溶液中添加定量金属络合剂(10 mmol/L EDTA-2Na 溶液)后,对乙腈、乙腈∶水(4∶1,体积比)、乙腈(含1%乙酸)、乙腈∶水(4∶1,体积比,含1%乙酸)、乙腈∶甲醇∶水(3∶1∶1,体积比)、乙腈∶甲醇∶水(3∶1∶1,体积比,含1%乙酸)6 种混合提取溶液的提取效果进行比较,同时对C18固相萃取柱和Oasis HLB 固相萃取柱的净化效果进行对比。
1.6 方法学验证试验
利用空白样品加标试验,对方法选择性、线性、基质效应(matrix effects,ME)、回收率、精密度、检出限(limit of detection,LOD)和定量限(limit of quantitation,LOQ)等参数进行测定评价。其中,允许使用兽药的最高残留限量(maximum residue limit,MRL)和禁用兽药分析方法的最低要求执行限量(minimum required performance limit,MRPL)数据来源于GB 31650—2019《食品安全国家标准食品中兽药最大残留限量》[8]、欧盟委员会法规、国际药典、美国联邦法案、日本《食品中农业化学品残留肯定列表制度》(简称《肯定列表》)[9-12]中的相关规定。对于目标兽药化合物中没有在法规中对水产品中残留量进行限定的物质进行方法学验证试验时,将其MRL 值默认为10 μg/kg。
1.6.1 化合物定性
依据保留时间(retention time,RT)以及母离子和子离子精确质荷比对目标兽药化合物进行定性分析。
1.6.2 线性和基质效应
配制浓度为1 000 μg/kg 的目标兽药化合物混合标准溶液,逐级稀释。选择0、0.5、1、2、5、10、20、50、100、200 μg/kg 10 个不同的添加浓度,在最适仪器条件下,进行UPLC-HRMS 上机试验。绘制目标化合物溶剂标准曲线,计算相应的线性回归方程及相关系数。同时通过比较目标化合物的溶剂和基质匹配标准曲线,对其基质效应进行评价,计算公式如下。
X=(Sa-Sb)/Sb×100
式中:X 为基质效应,%;Sa为基质标准曲线斜率;Sb为溶剂标准曲线斜率。
1.6.3 检出限和定量限
采用标准曲线匹配法和逐级稀释法向空白样品中添加已知浓度的标准溶液,选择0.2、0.4、0.5、0.8、1.0、2.0、4.0、10.0 μg/kg 8 个添加浓度水平。在最适仪器条件下,进行UPLC-HRMS(Orbitrap)上机试验,以3 倍和10 倍信噪比(signal/noise,S/N)分别计算方法的LOD 和LOQ。
1.6.4 加标回收试验
向长尾鳕、南美白对虾、栉孔扇贝水产样品中分别添加由79 种兽药标准溶液配制成的混合标准溶液,根据国内外法规相关规定及计算所得LOQ,确定79种目标兽药化合物添加浓度(氯霉素类为0.3 μg/kg,三苯甲烷染料类、非甾体雌激素类为2 μg/kg,喹诺酮类、磺胺及甲氧苄啶类为5 μg/kg,其余化合物为10 μg/kg)。在1 倍、2 倍和10 倍3 种添加浓度条件下各重复测定6 次,计算回收率及相对标准偏差(relative standard deviation,RSD)。
2 结果与分析
2.1 质谱条件优化
本试验的兽药化合物种类多,为便于统计依据物质结构将79 种兽药分为11 大类,即苯并咪唑类(6 种)、β-内酰胺类(11 种)、糖皮质激素类(8 种)、林可酰胺类(2 种)、大环内酯类(3 种),喹诺酮类(18 种)、磺胺及甲氧苄啶类(21 种)、四环素类(4 种)、三苯甲烷染料类(2种)、酰胺醇类(3 种)和非甾体雌激素类(1 种)。
首先确定79 种目标兽药化合物相应的中英文名称、分子式等基本信息,根据得到的分子式对其理论分子质量做初步推断。利用自动进样器将各个目标兽药化合物分别直接注入高分辨质谱,采用全扫描/数据依赖二级扫描(Full MS/dd-MS2)模式,对目标兽药化合物标准溶液进行质谱检测,得到一级全扫描质谱图。依据一级质谱图确定各个目标化合物的母离子精确质量数,将实际测得的母离子精确质量数与理论值进行对比后,确定目标化合物的最优母离子加合形式。目标化合物在不同归一化碰撞能量条件下采集得到二级质谱图,从各个目标化合物的二级质谱图中选择响应值较高的3~5 个碎片离子,并记录下其相应的实测子离子质荷比。利用Xcalibur 4.0 软件对得到的碎片离子的元素组成进行拟合(在误差为5×10-6的条件下,确定每个碎片离子最小偏差时的元素组成),根据不同元素组成中的同位素质荷比和元素组成相对丰度,计算出子离子理论精确质量。建立满足试验要求的79 种目标化合物的高分辨质谱库,部分代表目标兽药化合物的试验结果如表1 所示。
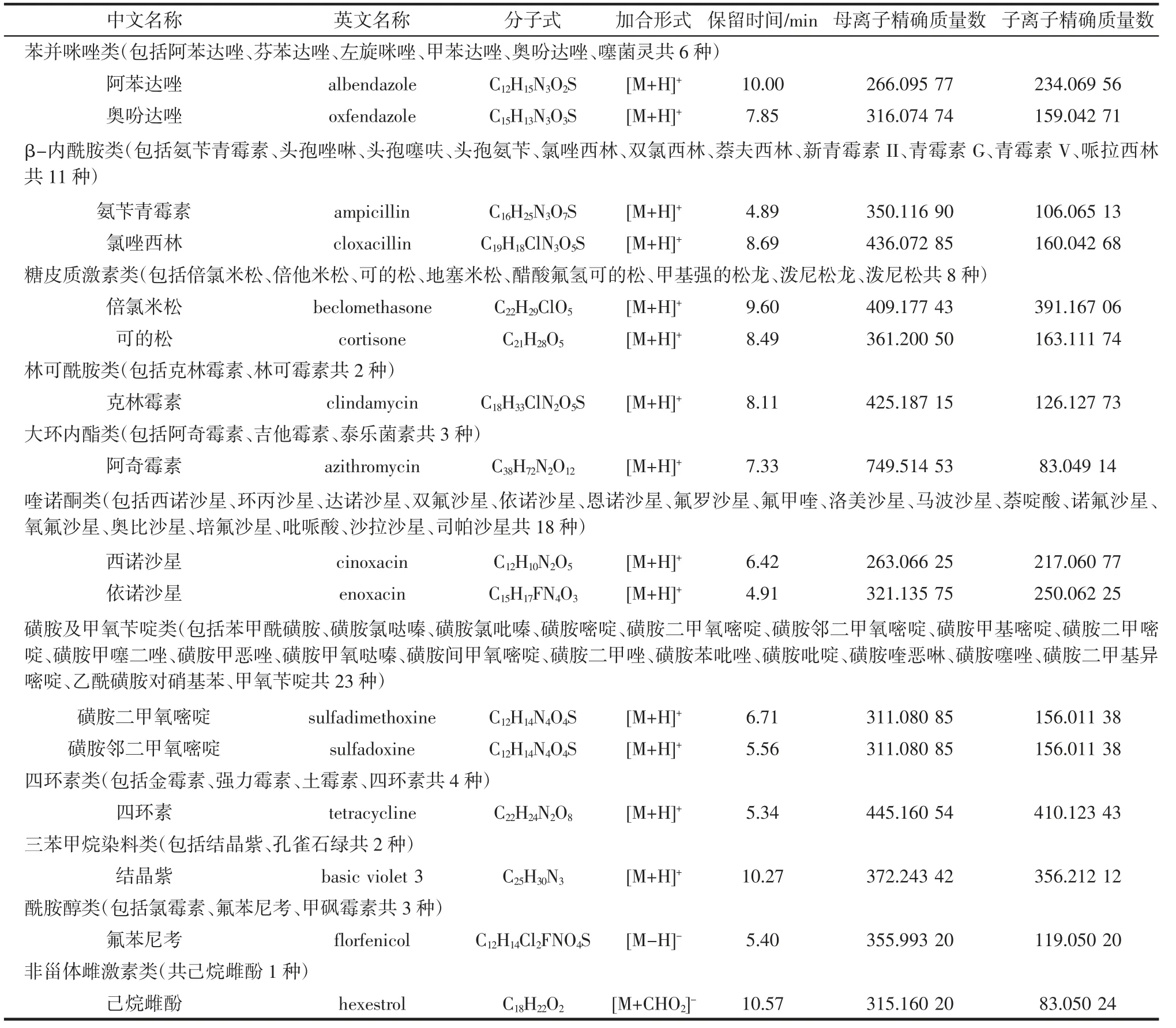
表1 11 类兽药代表目标化合物基本信息及各项UHPLC-HRMS 参数信息Table 1 Basic information and UHPLC-HRMS parameters on representative target compounds of 11 categories of veterinary drugs
2.2 色谱条件优化
2.2.1 流动相的优化
流动相可能会影响目标化合物的保留时间和峰形,并对定量分析结果造成影响[18]。不同流动相对代表化合物响应强度的影响如图1 所示。
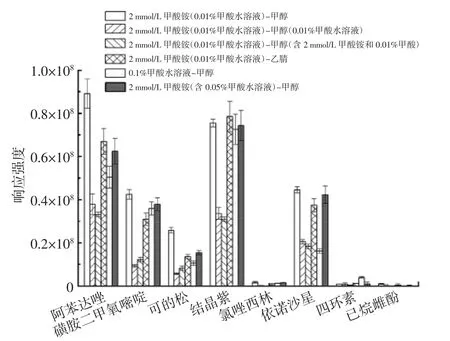
图1 代表化合物在不同流动相条件下的响应强度Fig.1 Response intensity of representative compounds under different mobile phase conditions
由图1 可知,在正离子模式下,0.1%甲酸水溶液作为流动相,可有效促进化合物的离子化。但在负离子模式下,甲酸的加入不利于去质子化作用和化合物的电离,对己烷雌酚的抑制作用十分明显。为了在一次上机试验内对正负离子模式同时进行检测以得到优良的试验结果,向流动相内添加一定量的甲酸铵,减少甲酸的添加量。当水相为2 mmol/L 甲酸铵(含0.01%甲酸)水溶液时,可保证所有化合物均具有较好的峰形和响应强度。对于绝大多数目标化合物而言,有机相为甲醇时的响应强度略高于有机相为乙腈时的响应强度。从试验结果和节约成本等角度综合考虑,最终确定流动相为2 mmol/L 甲酸铵(含0.01%甲酸水溶液)-甲醇。
2.2.2 色谱柱的选择、进样量及洗脱梯度的确定
对于兽药化合物,反相色谱柱具有较好的分离效果,经查阅资料后和试验发现[17-18],Acquity UPLC BEH C18色谱柱(100 mm×2.1 mm×1.7 μm)是一类对绝大多数兽药化合物都具有良好的分离效果且能够得到清晰尖锐色谱峰形,适用于大多数兽药检测的反向色谱柱。因此本研究确定Acquity UPLC BEH C18色谱柱为试验色谱柱。
反相色谱柱极性化合物优先流出,增加有机相在流动相中所占的比例,能够促进弱极性化合物的流出。当有机相的占比逐步提高到一定浓度后,保持该占比同时稳定一段时间,可将检测过程中样品内存在的非极性杂质送出,对色谱柱起到清洁和保护作用[19]。目标兽药化合物的分离试验结果如图2 和图3 所示。

图2 兽药化合物目标物的分离结果(正离子模式)Fig.2 Results of the separation of target compounds of veterinary drugs(positive ion mode)
图3 兽药化合物目标物的分离结果(负离子模式)Fig.3 Results of the separation of target compounds of veterinary drugs(negative ion mode)
根据图2 和图3 的试验结果,对目标兽药化合物的洗脱分离情况进行分析可以发现,所有目标物分布在3.29 min(磺胺嘧啶)到10.68 min(芬苯达唑)之间,在整个洗脱20 min 内整体分离情况较为理想。
2.3 前处理条件优化
2.3.1 样品提取条件优化
水产品是一类基质复杂的动物源食品,前处理是建立多兽药同时检测分析方法中十分关键的一步。对目标样品进行前处理时,提取溶剂的选择对试验结果有着重要影响[20-22]。甲醇和乙腈是常用的提取溶剂。由于乙腈具有分散基质、沉淀蛋白、脂质在其中的溶解性较差等性能,可以降低蛋白质、脂质等对检测造成的影响,适用于水产品基质,考虑以乙腈作为初始提取溶剂,并对其进行优化[23-24]。水有助于提升样品中极性化合物的提取效率,乙酸有助于调节提取溶剂的pH值和对样品进行酸解。同时,由于目标兽药化合物中的四环素类药物易与溶液中的金属离子发生络合,需向提取液中加入一定量的金属络合剂。查阅资料后发现[17-18],可通过向提取溶液中加入一定比例的甲醇,来提高林可酰胺类和四环素类的提取效率。本研究根据不同有机溶液的添加比例确定了6 种不同的提取溶剂,并以回收率为指标,对这6 种提取溶液进行评价。在使用不同提取溶液的条件下,代表兽药化合物回收率的试验结果如图4 所示。

图4 代表化合物在不同提取溶剂检出回收率Fig.4 Recoveries of representative compounds detected by different extraction solvents
对图4 中目标化合物的回收率分析比较后发现,单独使用乙腈作为提取溶剂,对β-内酰胺类、喹诺酮类、磺胺类、三苯甲烷染料类、四环素类等绝大多数目标兽药化合物的提取效果较差,目标兽药化合物回收率较低,有超过73%兽药化合物的回收率小于50%。而当向乙腈中添加一定量的水后,用乙腈∶水(4∶1,体积比)作为提取溶剂时,β-内酰胺类、糖皮质激素类、大环内酯类、喹诺酮类、磺胺类等化合物的回收率有明显的提升,有超过87%化合物的回收率介于50%~120%,该提取溶剂对大多数目标兽药化合物具有较好的提取效果。一定量乙酸的加入提高了喹诺酮类化合物回收率的同时对磺胺类化合物的回收率有一定的负面影响,不过磺胺类化合物的回收率也能保证在70%左右;EDTA-2Na 溶液的加入有助于提高四环素类化合物的提取效率;而甲醇的加入则有助于提高林可酰胺类化合物和磺胺类化合物的提取效率。当使用乙腈∶水(4∶1,体积比)作为提取溶剂时,存在于70%~120%这个区间内的化合物数目更多;当使用乙腈∶甲醇∶水(3∶1∶1,体积比)作为提取溶剂时,磺胺类等化合物具有更高的回收率;而当使用乙腈∶甲醇∶水(3∶1∶1,体积比,含1%乙酸)作为提取溶剂时,可以保证包括喹诺酮类、四环素类等化合物具有更高的回收率。因此综合考虑之后,本研究最终确定乙腈∶甲醇∶水(3∶1∶1,体积比,内含1%乙酸)和10 mmol/L EDTA-2Na 溶液作为最终的提取溶剂。
2.3.2 净化条件优化
固相萃取法(solid phase extraction,SPE)[25-26]是将传统的液固萃取法和现代新兴液相色谱技术相融合而发展出来的前处理方式,也是经常被应用于兽药前处理的方法之一。C18固相萃取柱和Oasis HLB 固相萃取柱是常见的固相萃取柱,其中C18固相萃取柱的填料为十八烷基硅烷键合硅胶,可有效除去脂类、糖类等亲脂性杂质;Oasis HLB 固相萃取柱的填料为亲脂性二乙烯苯和亲水性N-乙烯基吡咯烷酮两种单体按一定比例聚合成的大孔共聚物,同时具有亲水性和疏水性基团,扩展了适用的pH 值范围和化合物的极性范围,对弱极性和许多中、强极性的化合物都具有很好的萃取效果[27-29]。两种固相萃取柱针对部分代表化合物的净化效果如图5 所示。
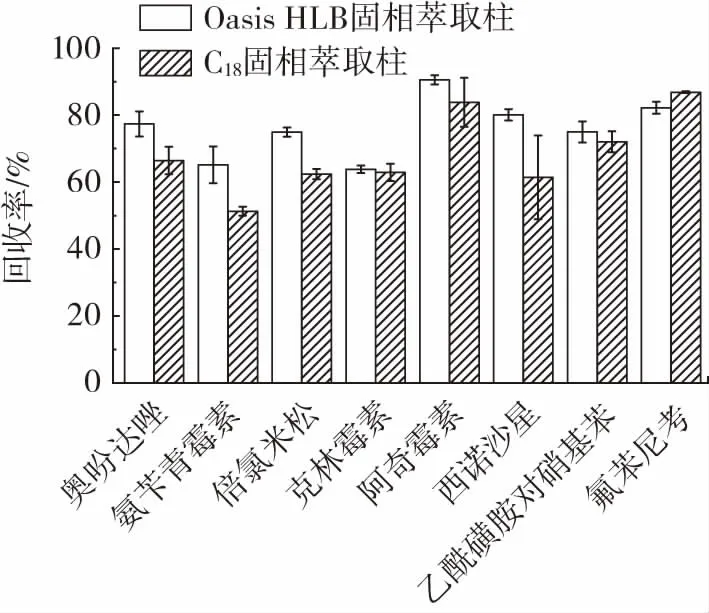
图5 不同净化方法条件下代表化合物回收率Fig.5 Recoveries of representative compounds by different purification methods
从图5 中可以看出,经Oasis HLB 固相萃取柱净化后,目标兽药化合物回收率较高且较多化合物的回收率集中在70%~120%,显示出更好的净化效果和回收率。因此,选择Oasis HLB 固相萃取柱作为本试验前处理的净化材料。
2.4 方法学验证
当保留时间、母离子精确质量数和响应值最强的定性子离子精确质量数在误差范围内符合相应条件时,就可以确认该样品为阳性检出样品。各目标物的定性参数见表1。对于目标物中存在的同分异构体,可以通过对不同的碎片离子质量数、保留时间、色谱峰等逐一进行比较,对各个目标化合物进行区分。此外,在试验过程中,发现各个目标物的色谱峰附近未有任何相关的基质干扰峰的出现,该仪器方法对于目标兽药化合物的分析检测具有较高的准确性。
2.4.1 线性关系和基质效应
依据UPLC-HRMS 试验结果和对所有目标兽药化合物在0.5~200.0 μg/kg 范围内绘制的目标化合物溶剂标准曲线以及相应的线性回归方程分析可以发现,所有目标化合物在0.5~200.0 μg/kg 内的溶剂标准中具有良好的线性关系,相关系数R2均大于0.99。
水产品基质复杂,在使用HESI 作为离子源的时候,当基质物质与目标物共存在于溶剂中且同时流出时,溶剂中所含有的复杂物质会共同在离子源区域发生离子化,当基质因子在-20%~20%之间时认为无明显基质效应的干扰,小于-20%表明基质抑制,大于20%表明基质增强。研究采用向长尾鳕、南美白对虾、栉孔扇贝水产样品提取液中加入相应的标准溶液,通过比较溶剂线性曲线和基质线性曲线,可以发现金霉素、强力霉素、土霉素3 种四环素类化合物在3 种基质内表现出明显的基质增强效应,其他各类化合物在3 种基质中均表现为基质抑制效应或不表现基质效应。
2.4.2 检出限和定量限
根据仪器在最适条件下,对含有目标兽药化合物的混合标准溶液试验测定所得的3 倍信噪比(S/N=3)和10 倍信噪比(S/N=10)计算,可确定该方法中各个目标兽药化合物的检出限和定量限。结果发现,在水产样品中79 种目标兽药化合的方法检出限范围为0.2~5.0 μg/kg,方法定量限范围为0.5~10.0 μg/kg。
2.4.3 方法准确度和精密度
对比长尾鳕、南美白对虾、栉孔扇贝3 种样品在1倍、2 倍和10 倍3 种添加浓度条件下的回收率,代表兽药化合物回收率的试验结果如图6~图8 所示。
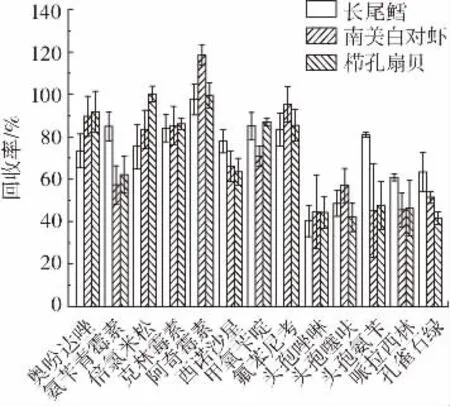
图6 在1 倍添加浓度条件下长尾鳕、南美白对虾、栉孔扇贝中代表化合物的回收率Fig.6 Recoveries of the representative compounds in Macrouroidei,Litopenaeus Vannamei,and Azumapecten farreri under the condition of once MRL

图7 在2 倍添加浓度条件下长尾鳕、南美白对虾、栉孔扇贝中代表化合物的回收率Fig.7 Recoveries of the representative compounds in Macrouroidei,Litopenaeus Vannamei,and Azumapecten farreri under the condition of twice MRL
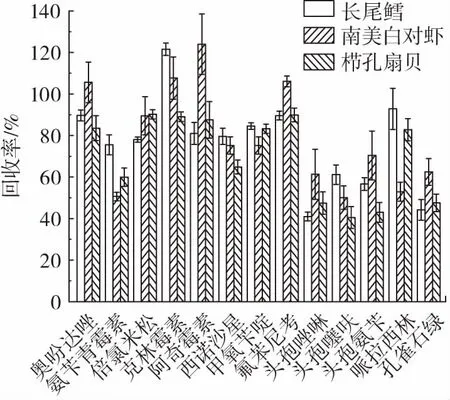
图8 在10 倍添加浓度条件下长尾鳕、南美白对虾、栉孔扇贝中代表化合物的回收率Fig.8 Recoveries of the representative compounds in Macrouroidei,Litopenaeus Vannamei,and Azumapecten farreri under the condition of 10 times MRL
从图6~图8 中可以看出,该方法在长尾鳕样品中的回收率为40.32%~122.12%;在南美白对虾样品中的回收率为40.70%~123.98%;在栉孔扇贝样品中回收率为40.41%~118.07%。在1 倍添加浓度条件下,鳕鱼、虾、扇贝样品中分别有96.2%、94.9%、93.7%兽药化合物的回收率在集中50%~120%,其中鳕鱼样品中有2种兽药(头孢唑啉和头孢噻呋)回收率小于50%,虾样品中有3 种兽药(头孢唑啉、头孢氨苄和哌拉西林)回收率小于50%,扇贝样品中有5 种兽药(头孢唑啉、头孢噻呋、头孢氨苄、哌拉西林和孔雀石绿)回收率小于50%,鳕鱼样品中的兽药化合物回收率略优于其余2 种;在2 倍添加浓度条件下,鳕鱼、虾、扇贝样品中分别有93.7%、92.4%、92.4%兽药化合物的回收率在集中50%~120%,然而虾样品中有84.8%兽药化合物的回收率集中于70%~120%,而扇贝样品中仅有46.8%兽药化合物的回收率集中于70%~120%;在10 倍添加浓度条件下,鳕鱼、虾、扇贝样品中分别有94.9%、93.7%、89.9%兽药化合物的回收率在集中50%~120%。因此该方法更加适用于鳕鱼、虾类等低脂肪、低蛋白质的水产样品检测,而对于扇贝这类同样低脂低蛋白但含有丰富色素类的水产品,该方法的回收率偏低,可能需要采用净化方法针对色素做进一步的优化,同时目标化合物在3 种样品中回收率的相对标准偏差均小于20%。查阅相关规定后可以发现,对于包括GB 31650—2019、欧盟委员会法规、“肯定列表制度”等相关国内国际法规中规定了在鱼类、虾类等水产品中最高添加限量相关要求的兽药化合物,本方法中这些目标兽药化合物的定量限低于其相关要求。该方法简单快捷,具有较好的准确性和灵敏度,能够适应不同水产品中兽药多残留的同时快速检测。
3 结论
基于超高效液相色谱-高分辨质谱仪同时检测水产品中多兽药残留,适宜用2 mmol/L 甲酸铵(含0.01%甲酸水溶液)-甲醇作为检测流动相;用乙腈∶甲醇∶水(3∶1∶1,体积比),并添加1%乙酸和10 mmol/L EDTA-2Na 作为提取溶剂;使用Oasis HLB 固相萃取柱进行净化。方法的最低检出限和定量限分别为0.2~5.0 μg/kg和0.5~10.0 μg/kg,水产品中除头孢唑啉、头孢噻呋、头孢氨苄、哌拉西林、孔雀石绿5 种化合物回收率较低外,其余回收率均在50%~120%,相对标准偏差为0.69%~19.91%。与现有标准方法相比,该方法的准确性和灵敏度较高,重现性好,定量限能够满足国标中针对鱼类规定的最大残留量快速筛查的相关要求,可以满足水产样品中79 种兽物化合物同时快速测定的需要。
