利用生物信息学筛选由2 型糖尿病肾病诱发肾小球炎症的关键基因
2023-11-14刘一卜彭金刚范菊娣
刘一卜,文 敏,彭金刚,范菊娣
(1.贵州医科大学药学院,贵州 贵阳 550004;2.贵州省化学合成药物研发利用工程技术研究中心,贵州 贵阳 550004)
糖尿病肾病(diabetic nephropathy,DN)是糖尿病中最为常见和严重的并发症,同时也是全世界终末期肾病最常见的原因[1]。截至2020 年,我国糖尿病合并慢性肾病患者数达2 430 万[1,2]。DN 发病机制复杂,目前尚无明确的诊断指标[3,4]。临床上筛查DN 主要针对尿白蛋白,然而这一指标并不灵敏[3-7]。因此,开发DN 诊断指标成为临床治疗的关键和难题。
DN 常伴随着肾小球、肾小管的损伤以及炎症反应[7]。DN 导致肾小球内皮细胞损伤主要有3 种方式:以多醇通路、蛋白酶C 为靶标影响细胞内信号传导和代谢;以合成晚期糖基化终产物(AGE)和氧化应激为标志诱导的肾小球细胞功能障碍和巨噬细胞的激活;由高糖等原因诱导的肾小球超滤和高血压[8,9]。肾小球内皮细胞损伤常将伴有炎症细胞(单核细胞、巨噬细胞、淋巴细胞)的富集和浸润,造成蛋白尿[10]。而低度肾小球炎症被认为是蛋白尿型肾小球病病程进展的共同途径,抗炎药物有利于控制DN 病程[11]。研究肾小球炎症的关键因子有助于发现DN 炎症新靶点、制定新策略。
20 世纪90 年代中期开始,人类基因组计划的完成,标志着进入了生物学驱动的时代[12]。随着分子、结构和化学生物学方法的发展,如基因组测序、微阵列基因表达分析、RNA 干扰(RNAi)、高通量结晶等方法[13],GEO[14]、GeneBank[15]、RefSeq[16]、Uni-Prot[17]数据库等可以帮助我们快速查找疾病相关的测序数据和蛋白序列信息,快速锁定疾病发生、发展的通路和相互作用网络,加速了疾病的临床和病理研究。
本文利用生物信息学,基于以往相关文献对GEO 数据集中的两组肾小球测序芯片数据GSE96804、GSE30122 进行分析,并交集出差异基因(DGEs),对差异基因进行GO 功能注释和GSEA-KEGG 通路富集,查找造成肾小球病变的关键促炎细胞因子及对应信号通路。通过PPI 网络锁定发病关键基因,联合公共数据库Nephroseq 探讨临床诊断DN 的关键诊断基因,为精确干预临床早期诊断、治疗提供指导和启发。
1 材料与方法
1.1 数据的处理和质量控制
在GEO 数据库(http://www.ncbi.nlm.nih.gov/geo)中按关键词“diabetes kidney glomeruli”进行检索,选择人源数据(Homo sapiens),同时选定测序方式为“Expression profilling by array”。在获得的数据中检查测序部位是否为肾小球,同时排除只含患病组或只含对照组的数据集,最终筛选得数据集GSE96804、GSE30122用于分析。基本信息见表1。

表1 数据集信息Tab 1 Dataset information
下载两组数据对应矩阵和平台文件,使用GEO2R 平台(https://www.ncbi.nlm.nih.gov/geo/geo2r/)分析数据的质量和差异。同时,基于平台注释文件,标注矩阵中的探针ID,将其转化为“ENTREZ_ID”,后转化为基因名用于后续分析。
1.2 DEGs 的筛选和功能注释
通过基于R 语言的limma 包分别对基因表达谱进行差异分析,以|log2FC|>0.379(FCs>1.3)和P<0.05 作为筛选条件筛选DEGs,利用基于R 的ggplot2 和pheatmap 进行可视化,分别绘制火山图和热图。同时,将得到的差异基因通过基因本体论(GO)和基因组百科全书(KEGG)进行富集,为筛选出具有统计学意义的生物过程(biological process,BP)、细胞成分(cellular component, CC)和分子功能(molecular function,MF),选用P<0.05 作为筛选条件。
同时,使用基因集富集分析法(GSEA,软件版本:4.3.2)重新富集可能被筛选阈值过滤的差异基因,使用Molecular Signature Datebase(2023)对KEGG 术语(c2.cp.kegg.v2023.1)以及生物学过程(c5.go.bp.v2023.1)进行注释分析,将|NES|>1,NOMp<0.05 以及FDR<0.25 的基因集视为具有显著差异进行富集。
1.3 蛋白-蛋白相互作用网络分析(protein-protein interaction,PPI)及核心基因的筛选
在线网站STRING(https://www.cn.string-db.org/)中键入DEGs 以预测蛋白之间相互作用,将交集所得的差异基因(上调、下调基因)构建的网络图导入Cytoscape 中,利用Cytohubba 插件,针对整个网络,利用MMC 算法,以最短路径显示并筛选出排名前十的DEGs 作为Hub 基因。
1.4 肾小球损伤的DN 临床特征分析
基于公共临床数据库Nephroseq 库(https://www.nephroseq.org),验证两组数据集交集后的核心基因在正常组和模型组之间是否具有表达差异。同时,基于Nephroseq 对于Hub 基因对临床特征:与肾小球滤过率(GFR)、24 小时蛋白尿、血清肌氨酸的影响进行比较和论述,用于筛选具有生物标志潜力的Hub 基因。
2 结果
2.1 数据质量分析和DEGs 的识别
数据库数据GSE96804、GSE30122 均符合芯片测序数据的质量控制要求,模型组和对照组之间可以较好区分,如下图1。GSE96804 通过limma 筛选后共获得1 235 个DEGs,其中上调基因417 个,下调基因818 个。GSE30122 筛选得DEGS 1 706 个,其中上调基因852 个,下调基因854 个。通过ggplot2和pheatmap 绘制的火山图和热图展示了DGES 的聚类和不同样本之间的差异,如图2 所示。

图1 芯片数据质量控制Fig 1 GSE96804 differential gene screening
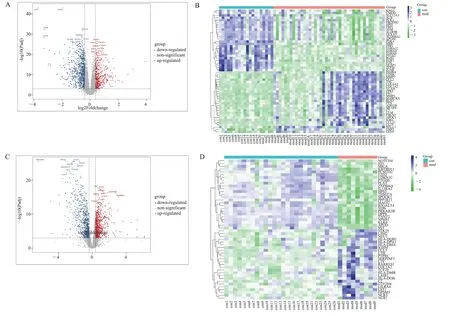
图2 GSE96804 差异基因筛选Fig 2 GSE96804 differential gene screening
2.2 富集结果分析
将两组筛选得的差异基因分别进行GO 富集分析,结果如图3。GO 富集结果涵盖了糖尿病肾病的主要生物学过程:外源性刺激、脂质水平的变化、免疫系统调节能力的激活、细胞趋化和分化以及肾脏的发育和代谢过程的异常[10,11]。与炎症因子相关的分子主要包括转化因子、促炎症因子以及相关黏附因子、趋化因子、Toll 样受体、脂肪因子、核受体等[3,18]。进一步利用GSEA 富集基因和通路,结果如图4。收集了包括转录因子(transcription factors)通路、Toll 样受体通路、脂肪细胞因子(adipocytokine)信号通路、细胞黏附(adherens)通路、细胞趋化因子(chemokine)通路,共6 个通路的基因富集结果。对二者富集到的DGEs 进行交集,共获得174个与炎症因子相关的DGEs。
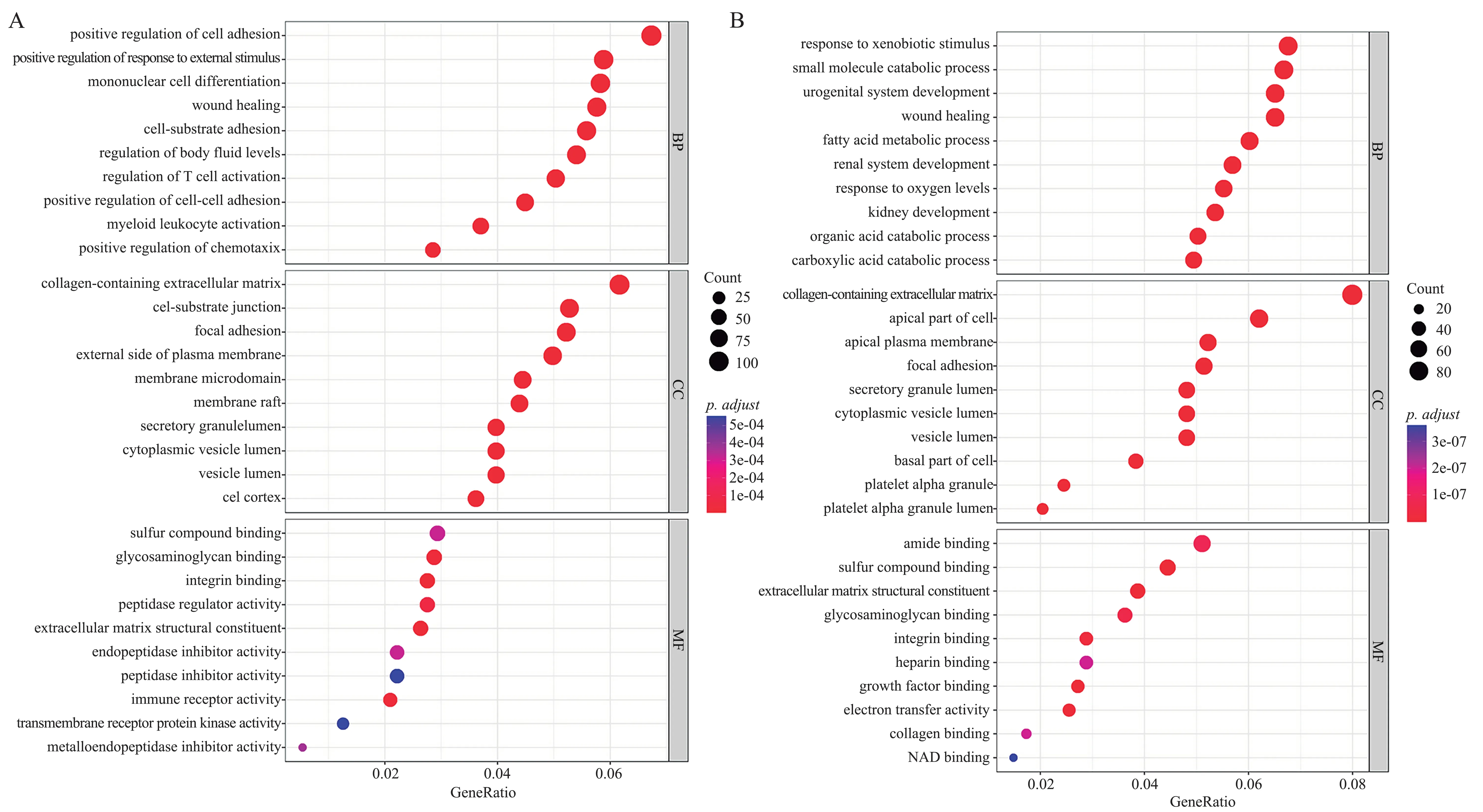
图3 GO 富集分析结果Fig 3 GO enrichment analysis results
2.3 核心基因的筛选和验证
使用String 数据库构建PPI 网络,将筛选得174个DGEs 输入到Cytoscape 中进行筛选,选取排名前10 位的DGEs 作为Hub 基因,结果如下图(图5)所示。

图5 与炎症相关通路排名前10 的关键基因Fig 5 Top 10 key genes associated with inflammatory pathways
将10 个Hub 基因输入到Nephroseq V5 数据库进行检索,检查是否是临床样本上具有表达差异的基因,结果如图6 所示。分析删除了在正常组与模型组没有表达差异的基因CD80,而TNF、CD4、CD80、CD86查询不到2 型糖尿病模型样本,而基因CD40LG查询不到肾小球部位的样本,故而不考虑。此处,筛选出具有表达差异的Hub 基因共5 个:CD8A、PTPRC、TLR2、CCL5、ITGAM。5 个基因在Neohroseq 数据集中的模型组(组2)与正常组(组1)表达量差异如图7 所示。5 种基因在模型组均上调,而除ITGAM外,其余4 种基因在正常组表达较少。

图6 Hub 基因在Nephroseq 数据库中表达差异分析Fig 6 Analysis of Hub's gene for differences in expression in the Nephroseq database
2.4 关键基因风险评估
通过检索Nephroseq V5 数据库,对比上述筛选所得Hub 基因:CD8A、ITGAM、PTPRC、CCL5、TLR2与蛋白尿、血清肌氨酸变化、肾小球滤过率等功能差异的相关性。对此,检索了正常组与模型组的肾脏样本进行差异分析,结果如图9~11。图8 展示了4 个Hub 基因表达量与肾小球滤过率(mL/min/1.73 m2)之间的相关性。CD8A、PTPRC、ITGAM、TLR2的高表达将导致GFR 偏低,引起肾损伤。图9 展示了3 个Hub 基因表达量与蛋白尿(g/24 h)之间的相关性。在CD8A与ITGAM高表达的条件下,蛋白尿将得到改善,而ITGAM基因则依赖较低水平的表达。图10 展示了Hub 基因表达量与血清肌氨酸(mg/dL)之间相关性。如图,除PTPRC外,ITGAM、CCL5、TLR2的上调将引起血清肌氨酸水平的上升。由此,5 种Hub 基因与肾病炎症确有相关性,可以作为具有临床潜力的生物标志物。

图8 Hubs 基因表达情况与GFR 相关性比较Fig 8 Comparison of Hubs gene expression and GFR correlation
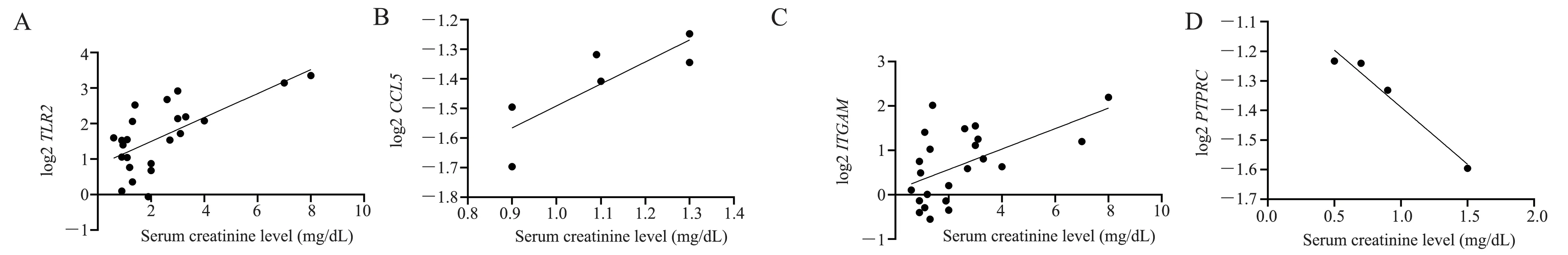
图10 Hubs 基因表达情况与血清肌氨酸相关性比较Fig 10 Comparison of Hubs gene expression with serum sarcosine
3 讨论
DN 成因相当复杂,与肾脏功能障碍和部分肾脏细胞的变化密切相关。多项研究已经证实,炎症相关因子和信号通路影响了DN 的发生和发展[18],筛选有关炎症因子可以帮助诊断和制定新的治疗方案。
本研究采用生物信息学的方法,选取两个人源肾小球细胞数据集进行分析和比对,探索引发肾小球炎症的关键基因。通过对两个GEO 生物芯片数据集进行交集分析,对比DN 患者和正常人的肾小球组织,通过GO、GSEA 进行注释,最终交集了174个与炎症相关的DGEs,同时通过Nephroseq 数据集进行验证并检查其在关键临床特征上的表现,最终获得了5 个潜在的临床生物标志物:CD8A、ITGAM、PTPRC、CCL5、TLR2。ITGAM 通路与巨噬细胞相关[19],巨噬细胞发生极化、浸润促使DN 炎症的发生[20]。CCL5 是一类关键的趋化因子,趋化因子和黏附因子的作用将使得单核细胞和淋巴细胞富集,引起DN[21]。酪氨酸蛋白磷酸酶C(PTPYC)是调节细胞内信号传导代谢的关键基因[22],同时也是调节T 细胞和B 细胞的重要调节因子,其表达差异与免疫调节和炎症因子相关。TLR2 是Toll 样受体2,其下游的NF-κB 是一类诱发蛋白尿的重要转录因子,诱导炎症因子的释放[23]。CD8A是属于CD8 链编码的一个抗原,通过诱导白细胞分化,辅助T 细胞受体识别抗原并活化T 细胞[24,25]。Shimokawa 等[26]的研究发现小鼠体内CD8 细胞活化T 细胞,将有效预防由链脲佐菌素诱导的小鼠罹患糖尿病。同时,通过与Nephroseq 进行比对,DN患者和正常组上述5 个基因表达具有差异,且蛋白尿、血清肌氨酸、肾小球滤过率等指标差异显著,具有临床诊断潜力。
综上所述,本研究通过生物信息学对DN 患者肾小球炎症差异基因进行分析,结果富集到174 个显著差异的基因,涉及外源性刺激、脂质水平的变化、免疫系统调节能力的激活、细胞趋化和分化以及肾脏的发育和代谢过程的异常等重要生物学过程。同时通过对炎症通路的筛选和比对,发现了五个具有生物标志物潜力的关键基因:CD8A、ITGAM、PTPRC、CCL5、TLR2。由于本研究基于公共数据集中不同批次的人源肾脏样本,样本的病程和研究部位详细情况未知,后续将加大样本量,进一步通过动物模型和qPCR 验证核心基因作为生物标志物的表达差异和后期的患者生存曲线,为进一步优化DN 引起的肾病提供新的诊断方案。
作者贡献度说明:
刘一卜:负责论文构思、调查研究和文章撰写;彭金刚:负责数据分析、基金资助;文敏:负责方法论的审核和文章校对;范菊娣:负责写作指导、项目管理和方法论指导。
所有作者声明不存在利益冲突关系。
