四氢异喹啉类PCSK9变构抑制剂的合成
2023-11-10李文雅刘洪涛马志欣李文燕
李文雅, 刘洪涛, 马志欣, 李文燕
(1.河北师范大学 化学与材料科学学院,河北省有机功能分子重点实验室,河北 石家庄 050024;2.河北医科大学第一医院 药剂科,河北 石家庄 050000)
根据世界卫生组织(World Health Organization,WHO)报告,近年来心血管疾病(cardivascular disease,CVD)一直是全球死亡的首要原因[1-2].血液中低密度脂蛋白胆固醇(low density lipoprotein cholesterol,LDL-C)水平的升高是动脉粥样硬化性心血管疾病最主要的独立危险因素[3].因此,降低LDL-C是一种有效预防和治疗动脉粥样硬化性心血管疾病的策略[3].
前蛋白转化酶枯草溶菌素9(proprotein convertase subtilisin kexin type 9,PCSK9)是经过临床验证的有效治疗高胆固醇血症的药物靶点[4].2003年,Abifadel等[5]揭示了PCSK9在脂质代谢中的关键作用.在生理状态下,循环中的LDL-C与肝细胞表面的低密度脂蛋白受体(low density lipoprotein receptor,LDL-R)结合,然后内化到肝细胞中降解,LDL-R再循环到细胞表面,导致循环LDL-C水平下降[6-7].PCSK9通过与LDL-R结合并诱导LDL-R/PCSK9复合物进入溶酶体降解,阻止LDL-R的再循环[8].因此,抑制PCSK9是一种非常有效的降低LDL-C水平的方法.迄今已有3种PCSK9抑制剂用于临床,包括2015年由美国FDA批准的2种单克隆抗体(alirocumab和evolocumab)[9-11]和2020年美国FDA批准的1种小干扰RNA(inclisiran)[12-13].临床数据显示,它们在降低LDL-C水平方面优于他汀类药物[14].然而,作为大分子生物药物,高昂的治疗费用和注射给药限制了其广泛应用,降低了其长期优势[14].与之相比,小分子药物具有治疗成本更低、给药方式更多、药代动力学特性更优、免疫原性更低和副作用更少等优势.
小分子直接作用于PCSK9和LDL-R的结合域,阻止2者的蛋白-蛋白相互作用,无疑是最为有效的抑制方法.但是,PCSK9与LDL-R的结合域是一个平坦的疏水性区域,缺乏小分子稳定结合口袋,而直接抑制剂多为生物大分子,如单抗、多肽等[15].图1a给出的四氢异喹啉类化合物2-(1-甲基-6,7-二甲氧基-1,2,3,4-四氢异喹啉-1-基)-N-(噻唑-2-基)乙酰胺(1),是默克公司通过亲和质谱选择筛选(AS/MS)得到的一种变构抑制剂,晶体结构(PDB:6U3X)显示其结合部位在催化亚基与C端亚基之间,与精氨酸357、天冬氨酸360和精氨酸458发生相互作用,如图1b[16];药理实验也表明其可有效降低细胞内胆固醇水平,是一种良好的苗头化合物.为进一步开发此化合物,本文中,笔者对其合成进行了研究.

图1 化合物1的结构(a)及与PCSK9的结合(b)Fig.1 Structure of Compound 1(a) and Binding of Compound 1 to PCSK9(b)
化合物1的结构可以分为2-氨基噻唑、2-(3,4-二甲氧基苯基)乙胺和丁酰胺3部分.因此,以2-氨基噻唑和2,2,6-三甲基-4H-1,3-二噁英-4-酮为原料,二甲苯为溶剂,150 ℃下加热回流,酰化得到3-羰基-N-(噻唑-2-基)丁酰胺中间体a;将中间体a与3,4-二甲氧基苯乙胺加入到甲醇中,在50 ℃下加热回流发生缩合反应得到(E)-3-((3,4-二甲氧基苯乙基)氨基)-N-(噻唑-2-基)-2-丁烯酰胺中间体b;中间体b在H3PO4的催化作用下,加热至100 ℃进行反应,环合得到2-(1-甲基-6,7-二甲氧基-1,2,3,4-四氢异喹啉-1-基)-N-(噻唑-2-基)乙酰胺(1),合成路线如图2所示,总收率为79.4 %.
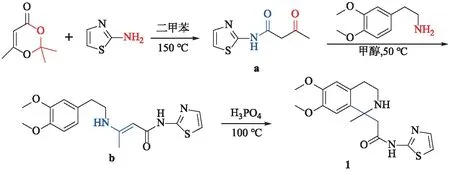
图2 化合物1的合成路线Fig.2 Synthesis Route of Compound 1
1 实验部分
1.1 主要仪器与试剂
薄层监测(TLC)采用2F-20D暗箱式紫外分析仪(巩义予华);质谱采用3200 QTRAP 1200 infinity series质谱仪(美国AB SCIESX公司);核磁共振采用WIPM-NMR-400核磁共振波谱仪(中科牛津波谱技术有限公司),四甲基硅烷(TMS)为内标.
化学试剂均为市售、分析纯.
1.2 实验步骤
1.2.1 3-羰基-N-(噻唑-2-基)丁酰胺的合成
将0.21 g 2-氨基噻唑(2.09 mmol)、0.28 mL 2,2,6-三甲基-4H-1,3-二噁英-4-酮(2.11 mmol)依次加入到0.50 mL二甲苯中,搅拌溶解,升温至150 ℃加热回流,TLC监测反应进程,30 min后反应完全.将反应液冷却至室温,有固体析出,减压抽滤,用无水乙醇冲洗滤饼得到褐色晶体0.34 g,产率为89 %,熔点为158.6~160.2 ℃.1HNMR(400 MHz,DMSO)δ:7.48(d,J=3.5 Hz,1H),7.23(t,J=4.5 Hz,1H),3.70(s,2H),2.20(s,3H).ESI-MS,m/z:185.2[M+H]+,207.4[M+Na]+.HR-MS:C7H8N2O2S for [M+H]+,calculated 184.030 6,found 184.030 1.
1.2.2 (E)-3-((3,4-二甲氧基苯乙基)氨基)-N-(噻唑-2-基)-2-丁烯酰胺的合成
向30 mL甲醇中依次加入0.50 g 3 -羰基-N-(噻唑-2-基)丁酰胺(2.71 mmol)和0.46 mL 3,4-二甲氧基苯乙胺(2.53 mmol),搅拌溶解后升温至50 ℃,过夜反应11 h.反应结束后溶液中有浅褐色固体析出,减压抽滤,用甲醇洗涤滤饼得到浅褐色固体0.88 g,产率为94 %,熔点为182.3~183.1 ℃.1HNMR(400 MHz,DMSO)δ:11.18(s,1H),9.13(t,J=5.8 Hz,1H),7.35(d,J=3.6 Hz,1H),7.00(d,J=3.6 Hz,1H),6.87(d,J=8.5 Hz,2H),6.79~6.74(m,1H),4.65(s,1H),3.75(s,3H),3.71(s,3H),3.42(q,J=6.7 Hz,2H),2.74(t,J=6.9 Hz,2H),1.85(s,3H).ESI-MS,m/z:348.3[M+H]+,370.4[M+Na]+.HR-MS:C17H21N3O3S for [M+H]+,calculated 347.130 4,found 347.129 8.
1.2.3 2-(1-甲基-6,7-二甲氧基-1,2,3,4-四氢异喹啉-1-基)-N-(噻唑-2-基)乙酰胺的合成
向3.02 mL H3PO4(30.81 mmol)中加入0.60 g (E)-3-((3,4-二甲氧基苯乙基)氨基)-N-(噻唑-2-基)-2-丁烯酰胺(1.73 mmol)搅拌溶解,升温至100 ℃进行反应,TLC监测反应进程,1 h后反应完全.向反应液中加入150 mL水淬灭,用CH2Cl2萃取2次,然后向水相中加入25 %的氨水溶液调节至pH=10,再用CH2Cl2萃取3次水相,用饱和NaCl溶液洗涤有机相后用无水硫酸镁干燥2 h,减压抽滤,滤液浓缩后进行柱层析分离(V(石油醚)∶V(乙醚)=1∶1)得到白色固体0.6 g,产率为95 %,熔点为156.8~157.6 ℃.1HNMR(400 MHz,DMSO)δ:7.40(s,1H),7.14(s,1H),6.82(s,1H),6.59(s,1H),3.69(d,J=11.7 Hz,6H),3.33(s,2H),3.01(dd,J=34.1,15.8 Hz,3H),2.81~2.59(m,3H),1.44(s,3H).13CNMR(101 MHz,DMSO)δ:169.62,157.75,147.55,147.53,138.04,133.44,127.19,113.65,112.34,110.10,56.21,55.74,55.13,46.43,38.30,29.51,29.25.ESI-MS,m/z:348.3[M+H]+,370.4[M+Na]+.HR-MS:C17H21N3O3S for [M+H]+,calculated 347.130 4,found 347.129 8.
2 结果与讨论
在环合得到目标化合物1的这一步反应中,尝试采用了CH3COOH,H3PO4,HCl 3种不同的酸作催化剂.采用CH3COOH时,反应转化率低;采用HCl时,反应生成的杂质较多;而采用H3PO4时,反应生成的杂质较少且易分离,产率高达95 %.因此,采用H3PO4作催化剂是反应条件的最优选择.
综上所述,以2-氨基噻唑和2,2,6-三甲基-4H-1,3-二噁英-4-酮为起始原料,经3步反应得到化合物1,总收率为79.4 %.此路线反应条件温和、操作简单、收率较高,为化合物1的进一步开发及其衍生物的合成奠定了基础,对新型治疗CVD药物的持续研究具有重要价值.
